Biochemistry Mastery
Biological Chemistry Fundamentals
Atoms, bonds, functional groups, thermodynamicsWater, pH & Biological Buffers
Water polarity, pH, Henderson-Hasselbalch, blood buffersAmino Acids & Protein Structure
Amino acid classes, peptide bonds, protein foldingEnzymes & Catalysis
Kinetics, Michaelis-Menten, inhibition, regulationCarbohydrates & Lipids
Sugars, glycogen, fatty acids, cholesterol, membranesMetabolism & Bioenergetics
ATP, glycolysis, gluconeogenesis, redox carriersCitric Acid Cycle & Oxidative Phosphorylation
Acetyl-CoA, ETC, ATP synthase, oxygen dependenceSignal Transduction & Cell Communication
GPCRs, kinases, calcium, hormone cascadesNucleic Acids & Gene Expression
DNA, replication, transcription, translation, epigeneticsBrain & Nervous System Biochemistry
Neurotransmitters, ion gradients, myelin, neurodegenerationHeart & Muscle Biochemistry
Cardiac metabolism, actin-myosin, energy systemsLiver Biochemistry
Glucose homeostasis, detox, urea cycle, bileKidney Biochemistry & Acid-Base
pH regulation, ion transport, hormonal functionsEndocrine System Biochemistry
Hormone classes, signaling, glucose & stress controlDigestive System Biochemistry
Gastric acid, enzymes, bile, absorption, microbiomeImmune System Biochemistry
Antibodies, cytokines, complement, oxidative burstAdipose Tissue & Energy Balance
Triglycerides, lipolysis, leptin, obesityTissue-Specific Metabolism
Fed vs fasting, organ fuel selection, starvationMolecular Basis of Disease
Diabetes, cancer metabolism, neurodegenerationClinical Biochemistry & Diagnostics
Blood tests, liver/kidney markers, lipid panelsRenal Architecture & Nephron Function
The kidneys are remarkable organs — each weighing only ~150 g but receiving 20–25% of cardiac output (~1.2 L/min). They filter ~180 L of plasma daily yet produce only ~1.5 L of urine, meaning 99% of filtered fluid is reabsorbed. Think of the kidney as a precision recycling plant: it first casts a wide net (filtration), then meticulously reclaims everything valuable while concentrating waste for disposal.
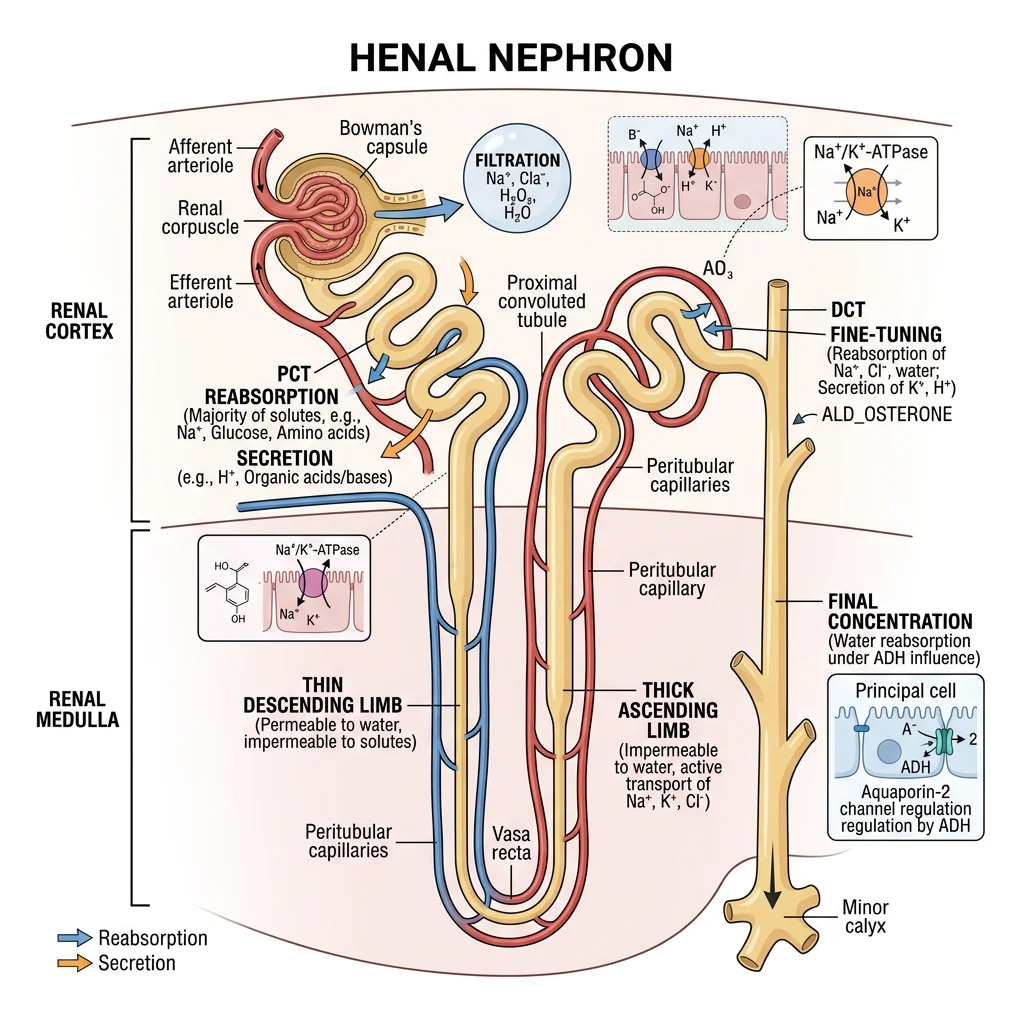
Kidney Organisation
- Cortex (outer): Contains all glomeruli, proximal convoluted tubules (PCT), distal convoluted tubules (DCT), and cortical collecting ducts. Receives ~90% of renal blood flow — high oxygen consumption for active reabsorption
- Medulla (inner): Contains loops of Henle, medullary collecting ducts, and vasa recta. Functions under hypoxic conditions — relies on anaerobic glycolysis. Creates the osmotic gradient (300 → 1200 mOsm/kg) essential for urine concentration
- Pelvis: Collects urine from collecting ducts → ureters → bladder
The Nephron — The Functional Unit
Each kidney contains ~1 million nephrons. Each nephron consists of:
- Glomerulus + Bowman's capsule: The filtration unit — fenestrated capillaries + podocytes create a three-layer filter (endothelium, basement membrane, podocyte slit diaphragm) that allows passage of water, electrolytes, glucose, amino acids, and small molecules while retaining proteins and blood cells
- Proximal convoluted tubule (PCT): The "workhorse" segment — reabsorbs ~65% of filtered Na⁺, water, glucose (SGLT2/SGLT1), amino acids, bicarbonate, and phosphate. Cells are packed with mitochondria and have a prominent brush border to maximise surface area
- Loop of Henle: Creates the medullary osmotic gradient via countercurrent multiplication — thin descending limb (water permeable, solute impermeable), thin ascending limb, and thick ascending limb (Na⁺-K⁺-2Cl⁻ cotransporter — NKCC2, target of loop diuretics like furosemide)
- Distal convoluted tubule (DCT): Fine-tunes Na⁺ and Ca²⁺ reabsorption — NCC (Na⁺-Cl⁻ cotransporter, target of thiazide diuretics), PTH-stimulated Ca²⁺ reabsorption via TRPV5
- Collecting duct: Final urine concentration — principal cells (ENaC for Na⁺ reabsorption, under aldosterone control; AQP2 for water reabsorption, under ADH control) and intercalated cells (H⁺ and HCO₃⁻ secretion for acid-base balance)
Two nephron types: Cortical nephrons (~85%, short loops) handle bulk reabsorption; juxtamedullary nephrons (~15%, long loops deep into medulla) are critical for urine concentration.
| Nephron Segment | Key Transporters | What's Reabsorbed | Clinical Drug Target |
|---|---|---|---|
| PCT | Na⁺/H⁺ exchanger (NHE3), SGLT2, Na⁺-phosphate cotransporter | 65% Na⁺/water, 100% glucose/amino acids, 80% HCO₃⁻, 65% K⁺ | SGLT2 inhibitors (empagliflozin, dapagliflozin) — block glucose reabsorption |
| Thick ascending limb | NKCC2 (Na⁺-K⁺-2Cl⁻), ROMK, paracellular Ca²⁺/Mg²⁺ | 25% Na⁺ (without water — "diluting segment") | Loop diuretics (furosemide) — block NKCC2 |
| DCT | NCC (Na⁺-Cl⁻), TRPV5 (Ca²⁺) | 5% Na⁺, Ca²⁺ (PTH-dependent) | Thiazide diuretics (hydrochlorothiazide) — block NCC |
| Collecting duct | ENaC (Na⁺), AQP2 (water), H⁺-ATPase | 3% Na⁺, variable water (ADH-dependent) | K⁺-sparing diuretics (amiloride — blocks ENaC; spironolactone — blocks aldosterone receptor) |
Glomerular Filtration & Tubular Reabsorption
The glomerulus acts as a high-pressure sieve — the unique arrangement of afferent and efferent arterioles (arteriole-to-arteriole, unlike any other capillary bed) maintains a hydrostatic pressure of ~55 mmHg, driving ultrafiltration through the three-layer barrier.
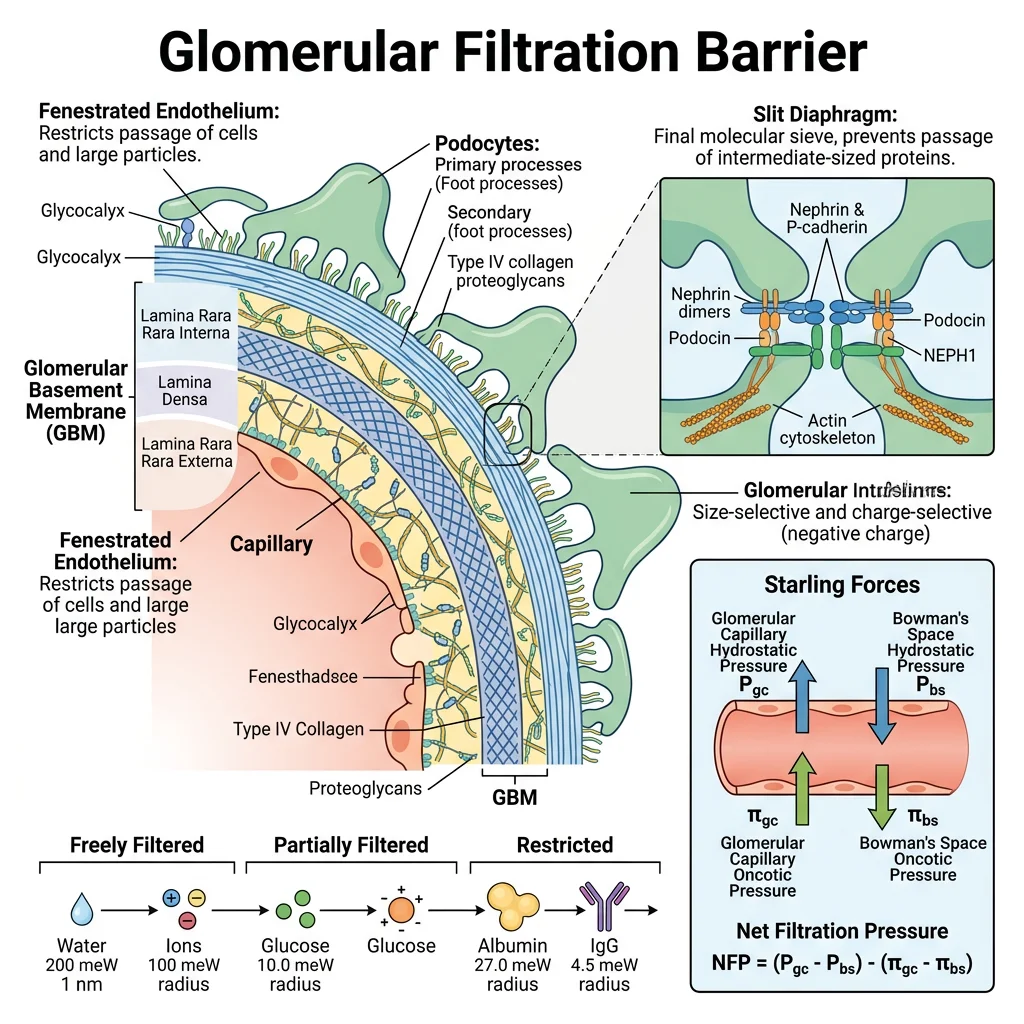
Glomerular Filtration Rate (GFR)
- Normal GFR: ~120 mL/min or ~180 L/day (adult). This means your entire plasma volume (~3 L) is filtered ~60 times daily
- Starling forces: GFR = Kf × (PGC − PBS − πGC + πBS), where PGC = glomerular capillary hydrostatic pressure (~55 mmHg), PBS = Bowman's space hydrostatic pressure (~15 mmHg), πGC = glomerular oncotic pressure (~30 mmHg), πBS ≈ 0. Net filtration pressure ≈ 10 mmHg
- Filtration barrier: Selectively filters by size and charge — freely filters molecules <7 kDa (water, glucose, urea, electrolytes); partially filters 7–70 kDa; blocks >70 kDa (albumin 69 kDa is largely retained). Negative charge of basement membrane further repels anionic proteins
- Autoregulation: GFR remains constant between MAP 80–180 mmHg via (1) myogenic response — afferent arteriole vasoconstriction and (2) tubuloglomerular feedback — macula densa senses NaCl delivery → adjusts afferent arteriolar tone via adenosine/ATP
SGLT2 Inhibitors — From Diabetes Drug to Multi-Organ Protector
The PCT reabsorbs 100% of filtered glucose (~180 g/day) via SGLT2 (90%, low affinity, high capacity) and SGLT1 (10%, high affinity, low capacity). SGLT2 inhibitors (empagliflozin, dapagliflozin, canagliflozin) block PCT glucose reabsorption → glycosuria → lower blood glucose.
But the benefits extend far beyond glucose lowering:
- Renoprotection: Increased NaCl delivery to macula densa → tubuloglomerular feedback → afferent arteriolar constriction → reduced glomerular hyperfiltration → slows CKD progression (DAPA-CKD trial, EMPA-KIDNEY trial)
- Cardioprotection: Reduced heart failure hospitalisations — possibly via natriuresis, osmotic diuresis, improved cardiac energetics (ketone body utilisation), and reduced cardiac fibrosis
- Now approved for CKD and heart failure regardless of diabetes status — a paradigm shift in nephrology and cardiology
Proteinuria — When the Filter Fails
Albuminuria (>30 mg/day) is the earliest sign of glomerular damage — clinically classified as:
- Microalbuminuria (30–300 mg/day): Early diabetic nephropathy, cardiovascular risk marker. ACE inhibitors/ARBs are first-line treatment (reduce glomerular pressure by dilating efferent arteriole)
- Macroalbuminuria (>300 mg/day): Established glomerular disease
- Nephrotic syndrome (>3.5 g/day): Massive proteinuria → hypoalbuminaemia → oedema → hyperlipidaemia. Causes: minimal change disease (children, podocyte foot process effacement), membranous nephropathy (adults, anti-PLA2R antibodies), diabetic nephropathy
Ion Transport & Electrolyte Balance
The kidney maintains the precise extracellular concentrations of Na⁺, K⁺, Ca²⁺, Mg²⁺, phosphate, and Cl⁻ — ion imbalances can be immediately life-threatening (cardiac arrhythmias from hyperkalaemia, seizures from hyponatraemia). The Na⁺/K⁺-ATPase on the basolateral membrane of every tubular cell is the engine that powers all renal transport — consuming ~6–8% of total body oxygen to maintain the electrochemical gradients that drive secondary active transport.
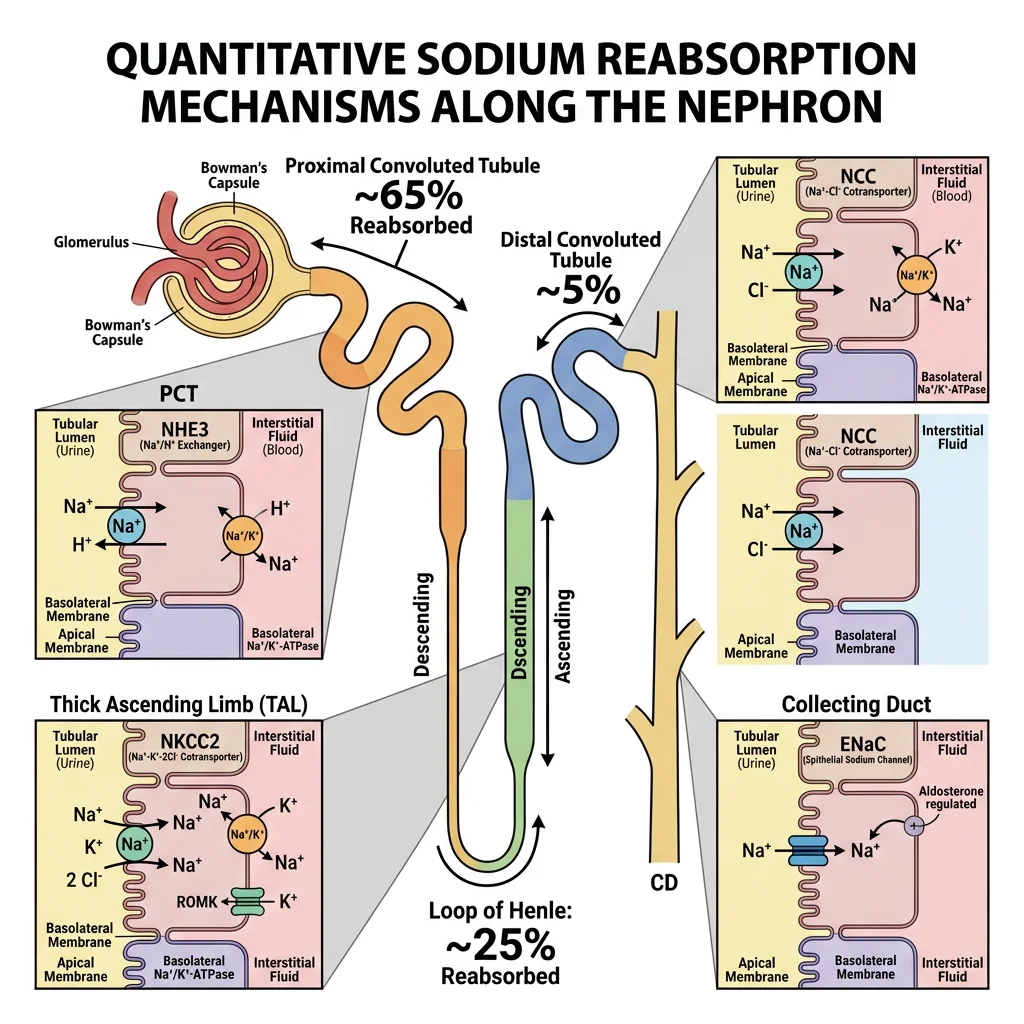
Sodium — The Master Ion
The kidney filters ~25,000 mmol Na⁺/day but excretes only ~150 mmol — 99.4% is reabsorbed. Sodium handling determines water balance, blood pressure, and the driving force for most other solute transport:
- PCT (65%): Na⁺/H⁺ exchanger (NHE3) on apical membrane — coupled to HCO₃⁻ reabsorption. Na⁺-glucose (SGLT2), Na⁺-amino acid, and Na⁺-phosphate cotransporters also reabsorb Na⁺. Water follows passively via aquaporin-1 (AQP1) — isotonic reabsorption
- Thick ascending limb (25%): NKCC2 (Na⁺-K⁺-2Cl⁻) — the "diluting segment" (water impermeable). Creates the corticomedullary osmotic gradient. Inhibited by furosemide
- DCT (5%): NCC (Na⁺-Cl⁻ cotransporter). Inhibited by thiazides
- Collecting duct (3%): ENaC (epithelial Na⁺ channel) — regulated by aldosterone (which increases ENaC and Na⁺/K⁺-ATPase expression). Inhibited by amiloride. Aldosterone interaction blocked by spironolactone/eplerenone
Potassium — The Dangerous Electrolyte
Serum K⁺ is maintained at 3.5–5.0 mmol/L — even small deviations cause cardiac arrhythmias because K⁺ sets the resting membrane potential of cardiomyocytes:
- Hypokalaemia (<3.5): Hyperpolarisation → muscle weakness, ileus, U waves on ECG, dangerous ventricular arrhythmias. Causes: vomiting (metabolic alkalosis drives K⁺ into cells), diarrhoea, loop/thiazide diuretics, Cushing syndrome
- Hyperkalaemia (>5.5): Depolarisation → peak T waves → widened QRS → sine wave → cardiac arrest. Causes: renal failure (reduced excretion), ACE inhibitors/ARBs, K⁺-sparing diuretics, rhabdomyolysis, acidosis (transcellular shift)
- Renal K⁺ handling: 65% reabsorbed in PCT (paracellular), 25% in thick ascending limb (ROMK recycling). K⁺ secretion occurs in principal cells of the collecting duct via ROMK channels — driven by aldosterone (which increases Na⁺ reabsorption → creates lumen-negative potential → drives K⁺ secretion)
| Electrolyte | Normal Range | Primary Renal Handling | Key Hormonal Regulator |
|---|---|---|---|
| Na⁺ | 135–145 mmol/L | 99.4% reabsorbed; fine-tuned in collecting duct (ENaC) | Aldosterone (↑ reabsorption), ANP (↓ reabsorption), ADH (water balance) |
| K⁺ | 3.5–5.0 mmol/L | 90% reabsorbed; secreted in collecting duct (ROMK) | Aldosterone (↑ secretion), insulin (↑ cellular uptake), pH |
| Ca²⁺ | 2.2–2.6 mmol/L (total) | 65% PCT paracellular, 25% thick ascending limb, 8% DCT (TRPV5 — active, PTH-stimulated) | PTH (↑ reabsorption in DCT, ↑ 1,25(OH)₂D), calcitonin (↓ reabsorption) |
| Phosphate | 0.8–1.5 mmol/L | 80% PCT reabsorption via NaPi-IIa/c cotransporters | PTH (↓ reabsorption — removes NaPi from membrane), FGF23 (↓ reabsorption) |
| Mg²⁺ | 0.7–1.0 mmol/L | 15% PCT, 70% thick ascending limb (paracellular via claudin-16), 10% DCT (TRPM6) | PTH (↑ reabsorption), loop diuretics (↑ excretion — cause hypomagnesaemia) |
Acid-Base Regulation
The body produces ~70 mmol of non-volatile acid daily (from protein metabolism, lactic acid, ketoacids) that cannot be exhaled as CO₂. The kidney is the only organ that can excrete this fixed acid load while simultaneously reclaiming the ~4,300 mmol of bicarbonate filtered daily. Blood pH must be maintained at 7.35–7.45 — a deceptively narrow range considering that pH is a logarithmic scale (pH 7.0 has 2.5× more H⁺ than pH 7.4).
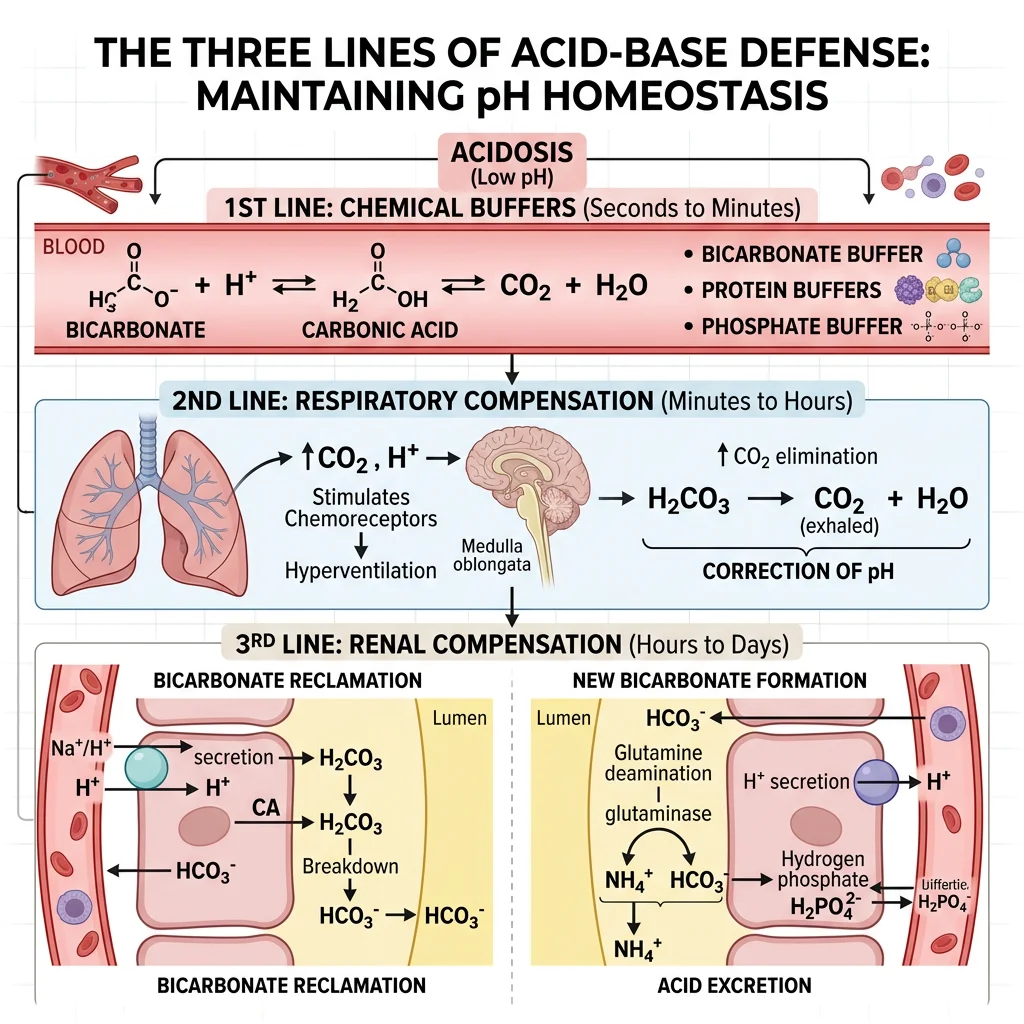
The Three Lines of Defence Against pH Change
- Chemical buffers (seconds): Bicarbonate (HCO₃⁻/CO₂, primary ECF buffer), haemoglobin, phosphate, plasma proteins — immediately neutralise added acid or base. The Henderson-Hasselbalch equation: pH = 6.1 + log([HCO₃⁻] / 0.03 × PCO₂)
- Respiratory compensation (minutes): Chemoreceptors detect pH change → adjust ventilation → change PCO₂. Acidosis → hyperventilation → ↓PCO₂; alkalosis → hypoventilation → ↑PCO₂. Fast but incomplete compensation
- Renal compensation (hours–days): The definitive defence — kidneys can both excrete acid AND generate new bicarbonate. Slower but more powerful — can fully correct pH disturbances that overwhelm buffers and lungs
Bicarbonate Reclamation
The kidney filters ~4,300 mmol HCO₃⁻ per day — losing even 5% would cause severe metabolic acidosis. The PCT reclaims ~80% of filtered bicarbonate, with the remainder reabsorbed in the thick ascending limb and collecting duct.
How Bicarbonate Reclamation Works
Bicarbonate itself is not directly reabsorbed — it requires an elegant indirect mechanism:
- Apical NHE3 (Na⁺/H⁺ exchanger) secretes H⁺ into the tubular lumen
- Luminal H⁺ + HCO₃⁻ → H₂CO₃ (carbonic acid) → catalysed by carbonic anhydrase IV (membrane-bound, luminal) → CO₂ + H₂O
- CO₂ diffuses freely into the PCT cell
- Intracellular carbonic anhydrase II converts CO₂ + H₂O → H₂CO₃ → H⁺ + HCO₃⁻
- H⁺ is recycled back to the lumen (step 1), while HCO₃⁻ exits via the basolateral Na⁺-HCO₃⁻ cotransporter (NBCe1) into blood
Net effect: For every H⁺ secreted into the lumen that combines with filtered HCO₃⁻, one "new" HCO₃⁻ is returned to the blood. No net acid excretion occurs — this is reclamation, not generation.
Carbonic Anhydrase Inhibitors — Acetazolamide
Acetazolamide inhibits carbonic anhydrase → blocks luminal conversion of H₂CO₃ → CO₂ + H₂O → impairs bicarbonate reclamation → HCO₃⁻ is lost in urine → metabolic acidosis (mild, self-limiting because as plasma HCO₃⁻ drops, less is filtered, and the effect diminishes).
Clinical uses: (1) Altitude sickness — induces metabolic acidosis → stimulates ventilation → improves oxygenation, (2) Glaucoma — reduces aqueous humour production, (3) Idiopathic intracranial hypertension — reduces CSF production. Also useful for metabolic alkalosis correction.
Ammonium Excretion
While bicarbonate reclamation prevents acid gain, net acid excretion requires generating NEW bicarbonate — achieved through ammonium (NH₄⁺) and titratable acid (H₂PO₄⁻) excretion. The ammonium system is particularly important because it can be upregulated 10-fold during chronic acidosis.
Renal Ammoniagenesis
- PCT glutaminase: Glutamine → glutamate + NH₄⁺; glutamate dehydrogenase → α-ketoglutarate + NH₄⁺. Each glutamine yields 2 NH₄⁺ and 2 HCO₃⁻ (from α-ketoglutarate metabolism)
- NH₄⁺ handling: Secreted into PCT lumen (substitutes for H⁺ on NHE3) → reabsorbed in thick ascending limb (substitutes for K⁺ on NKCC2) → concentrated in the medullary interstitium
- Collecting duct: NH₃ (uncharged) diffuses from medullary interstitium into acidic collecting duct lumen → combines with secreted H⁺ → trapped as NH₄⁺ (non-diffusible) → excreted in urine. This is "diffusion trapping"
- Adaptation: In chronic metabolic acidosis, PCT glutaminase expression increases (acidosis stabilises glutaminase mRNA) → up to 300 mmol/day NH₄⁺ excretion (vs normal ~40 mmol/day)
Titratable acid (~30 mmol/day): Filtered phosphate (HPO₄²⁻) buffers secreted H⁺ → H₂PO₄⁻ in urine. This contributes ~30% of net acid excretion under normal conditions.
| Acid-Base Disorder | pH | Primary Change | Compensation | Common Causes |
|---|---|---|---|---|
| Metabolic acidosis | ↓ | ↓ HCO₃⁻ | ↓ PCO₂ (hyperventilation) | DKA, lactic acidosis, renal failure, diarrhoea, RTA |
| Metabolic alkalosis | ↑ | ↑ HCO₃⁻ | ↑ PCO₂ (hypoventilation) | Vomiting, NG suction, loop diuretics, Cushing, Conn |
| Respiratory acidosis | ↓ | ↑ PCO₂ | ↑ HCO₃⁻ (renal, days) | COPD, opioids, neuromuscular disease, obesity hypoventilation |
| Respiratory alkalosis | ↑ | ↓ PCO₂ | ↓ HCO₃⁻ (renal, days) | Anxiety/hyperventilation, PE, salicylate poisoning, high altitude |
Hormonal Regulation
The kidney is both a target and a source of hormones. Three hormonal axes dominate renal regulation: ADH controls water reabsorption, aldosterone controls sodium and potassium balance, and ANP provides a counter-regulatory signal when blood volume is excessive. Together they orchestrate the body's fluid and electrolyte homeostasis with remarkable precision.
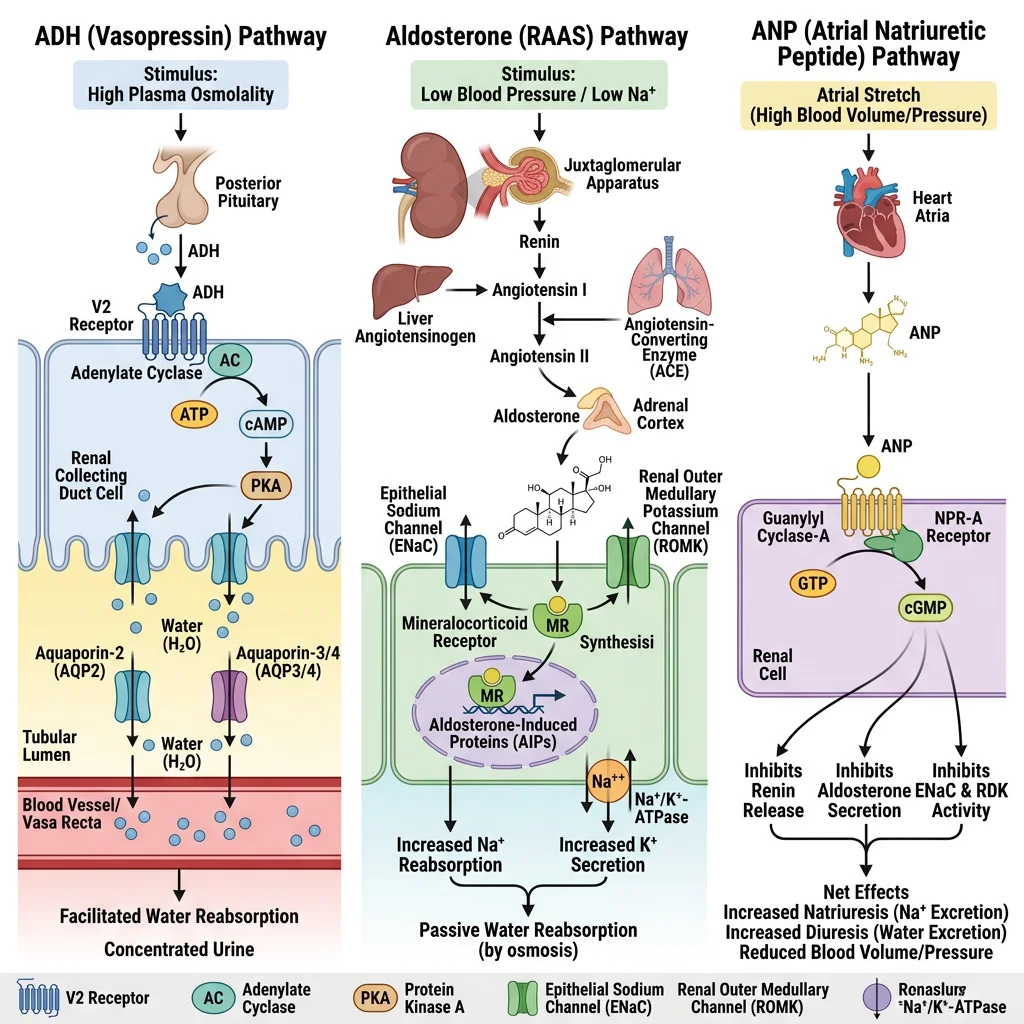
ADH (Vasopressin)
Antidiuretic hormone (ADH), also called vasopressin, is a 9-amino acid peptide synthesised in the hypothalamus (supraoptic and paraventricular nuclei) and released from the posterior pituitary. It is the master regulator of water balance.
ADH — Making Concentrated Urine
- Stimulus: ↑ plasma osmolality (>285 mOsm/kg, detected by hypothalamic osmoreceptors) or ↓ blood volume (>10% decrease detected by aortic/carotid baroreceptors). Osmolality is the primary driver under normal conditions
- Mechanism: ADH binds V2 receptors (basolateral) on collecting duct principal cells → Gs-coupled → ↑cAMP → PKA activates → AQP2 water channels inserted into apical membrane via vesicle fusion → water flows from tubular lumen → medullary interstitium (following the osmotic gradient) → vasa recta → blood
- Without ADH: Collecting duct is water-impermeable → dilute urine (up to 20 L/day in diabetes insipidus)
- With maximal ADH: Urine concentrated to ~1200 mOsm/kg (maximum = medullary interstitium osmolality)
- AQP2 regulation: Short-term — vesicle trafficking (minutes); long-term — ADH increases AQP2 gene transcription. AQP3 and AQP4 on the basolateral membrane are constitutively expressed
Diabetes Insipidus vs SIADH
- Central DI: Insufficient ADH production (post-pituitary surgery, trauma, tumours) → massive dilute polyuria (5–20 L/day), polydipsia, hypernatraemia. Treatment: desmopressin (DDAVP, synthetic ADH analogue)
- Nephrogenic DI: Kidney is resistant to ADH — AQP2 cannot be inserted. Causes: lithium therapy (most common drug cause — inhibits AQP2 expression), hypercalcaemia, hypokalaemia, congenital V2 receptor or AQP2 mutations. Treatment: thiazide diuretics (paradoxically reduce urine volume via enhanced proximal reabsorption), low-sodium diet, NSAIDs
- SIADH: Excessive ADH secretion → excessive water retention → dilutional hyponatraemia (Na⁺ <135, often <120 mmol/L) → cerebral oedema → confusion, seizures, coma. Causes: small cell lung cancer, CNS disorders, drugs (SSRIs, carbamazepine). Treatment: fluid restriction, hypertonic saline (if severe/symptoms), tolvaptan (V2 receptor antagonist — "vaptan")
Aldosterone
Aldosterone is a mineralocorticoid steroid hormone produced by the zona glomerulosa of the adrenal cortex, regulated by the renin-angiotensin-aldosterone system (RAAS) and directly by serum potassium levels.
The RAAS Cascade
- Trigger: ↓ renal perfusion pressure, ↓ NaCl at macula densa, or ↑ sympathetic stimulation → juxtaglomerular cells release renin
- Renin (protease) cleaves angiotensinogen (liver) → angiotensin I (inactive)
- ACE (angiotensin-converting enzyme, mainly in pulmonary endothelium) converts Ang I → angiotensin II (potent vasoconstrictor)
- Angiotensin II effects: (a) Vasoconstriction → ↑BP; (b) Stimulates aldosterone release; (c) Stimulates ADH release; (d) Stimulates thirst; (e) Preferentially constricts efferent arteriole → maintains GFR despite ↓ renal blood flow; (f) Enhances proximal Na⁺ reabsorption (NHE3 activation)
- Aldosterone enters collecting duct principal cells → binds intracellular mineralocorticoid receptor → ↑ ENaC expression (Na⁺ reabsorption) + ↑ ROMK expression (K⁺ secretion) + ↑ Na⁺/K⁺-ATPase expression
Clinical blockade: ACE inhibitors (ramipril, lisinopril) and ARBs (losartan, valsartan) are first-line antihypertensives — reduce both angiotensin II effects and aldosterone secretion. Side effects: hyperkalaemia (less K⁺ secretion), cough (ACEi — bradykinin accumulation), risk of AKI in bilateral renal artery stenosis (efferent arteriolar dilation → ↓GFR).
ANP (Atrial Natriuretic Peptide)
ANP is the counter-regulatory hormone to RAAS — released from atrial cardiomyocytes when atrial wall stretch increases (volume overload). BNP (brain natriuretic peptide) is released from ventricular cardiomyocytes under similar conditions.
ANP/BNP — The Volume-Reducing Hormones
- Renal effects: ↑ GFR (afferent arteriolar dilation + efferent constriction), ↑ Na⁺ excretion (natriuresis — inhibits ENaC and Na⁺ reabsorption in collecting duct), ↑ water excretion (diuresis)
- Suppress RAAS: Inhibit renin release, inhibit aldosterone secretion, inhibit ADH release
- Vascular: Vasodilation → ↓ systemic vascular resistance → ↓ blood pressure
- Mechanism: ANP binds NPR-A (natriuretic peptide receptor A) → guanylyl cyclase activation → ↑ cGMP → downstream effects
- Clinical biomarker: BNP and NT-proBNP are used to diagnose and monitor heart failure — elevated levels (>100 pg/mL for BNP) indicate cardiac volume/pressure overload. Sacubitril (neprilysin inhibitor) blocks ANP/BNP degradation → enhanced natriuretic effects — combined with valsartan (ARNI therapy) for heart failure with reduced ejection fraction
Kidney Disease Biomarkers
Assessing renal function relies on a panel of biomarkers. No single test captures glomerular, tubular, and interstitial function simultaneously, so clinicians combine several markers to build a complete picture. Understanding what each marker measures — and its limitations — is essential for correct diagnosis and staging of kidney disease.
| Biomarker | Source | Normal Range | What It Measures | Limitations |
|---|---|---|---|---|
| Serum Creatinine | Non-enzymatic breakdown of creatine phosphate in muscle | 60–110 µmol/L (0.7–1.3 mg/dL) | Inverse proxy for GFR — rises when GFR falls below ~50% | Affected by muscle mass (bodybuilders, amputees), diet (cooked meat), drugs (trimethoprim inhibits tubular secretion → false ↑). Not sensitive to early CKD ("creatinine-blind" range) |
| Blood Urea Nitrogen (BUN) | Hepatic urea cycle — end product of protein catabolism | 2.5–7.1 mmol/L (7–20 mg/dL) | Nitrogen waste clearance; BUN:creatinine ratio helps distinguish pre-renal (>20:1) vs intrinsic renal (~10:1) | ↑ by GI bleeding, high-protein diet, catabolic states, steroids — not specific for kidney disease alone |
| eGFR | Calculated from serum creatinine + age + sex (CKD-EPI 2021 equation) | >90 mL/min/1.73 m² | Best overall estimate of kidney function — used for CKD staging | Equations assume steady-state creatinine — inaccurate in AKI (rapidly changing). CKD-EPI 2021 removed race variable for equity |
| Cystatin C | Produced by all nucleated cells at constant rate | 0.6–1.0 mg/L | GFR marker independent of muscle mass — better for extremes (elderly, malnourished, amputees) | Affected by thyroid disease (↑ in hyperthyroidism), corticosteroids, inflammation. More expensive than creatinine |
| Urine ACR (Albumin-to-Creatinine Ratio) | Spot urine sample — normalises albumin to creatinine concentration | <3 mg/mmol (<30 mg/g) | Glomerular barrier integrity — detects early diabetic and hypertensive nephropathy | Transient ↑ with fever, exercise, UTI. Confirm with 2 of 3 positive samples over 3 months |
| KIM-1 / NGAL | Tubular injury markers (proximal tubule / neutrophils + tubular cells) | Not yet standardised | Early AKI detection — rise 12–24 hours before serum creatinine | Research stage to clinical translation. NGAL affected by sepsis, inflammation |
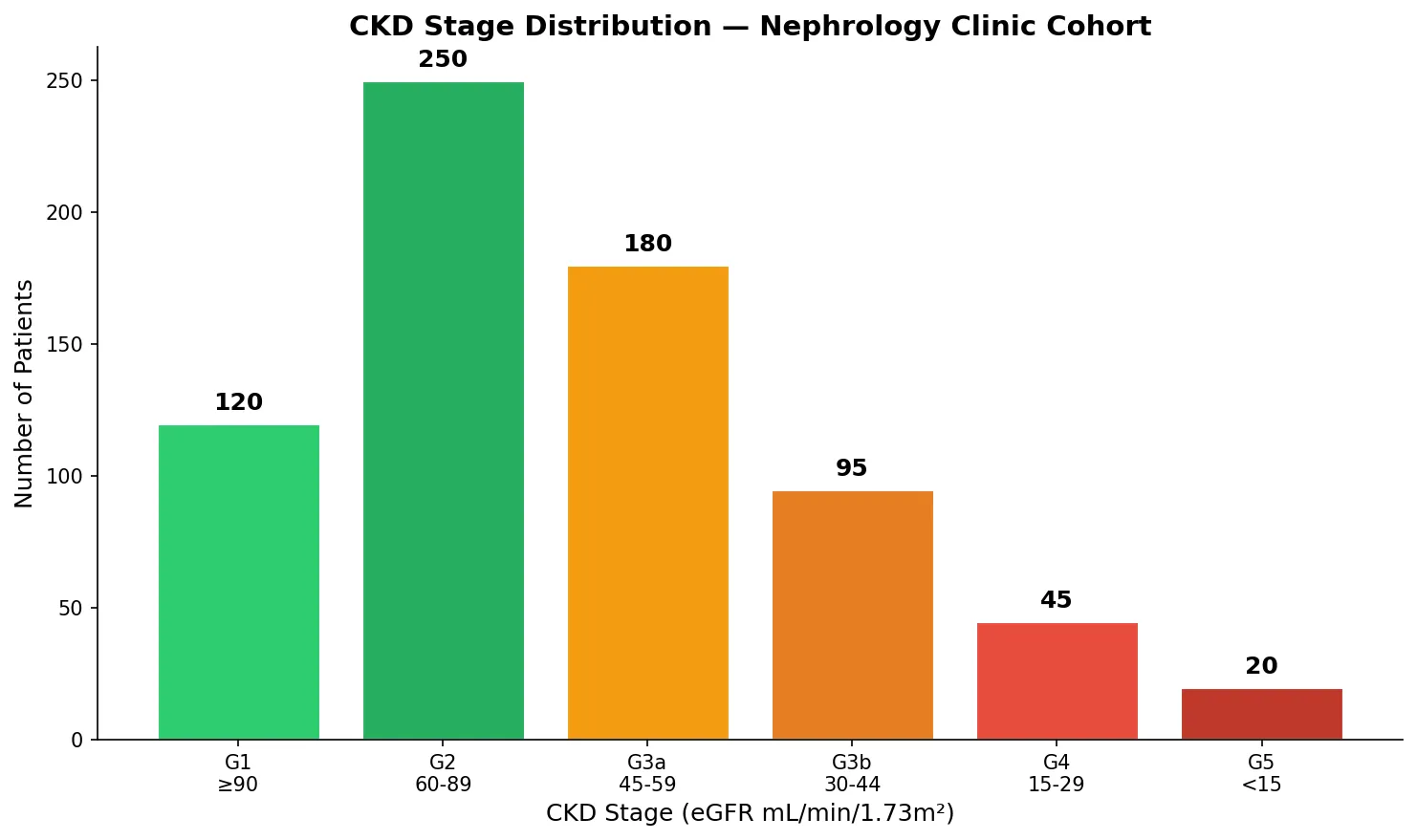
CKD Staging (KDIGO 2012)
Chronic kidney disease is staged by eGFR and albuminuria — both independently predict cardiovascular and renal outcomes:
- G1: eGFR ≥90 — normal or high (CKD only if albuminuria or structural abnormality present)
- G2: eGFR 60–89 — mildly decreased
- G3a: eGFR 45–59 — mild-to-moderate decrease
- G3b: eGFR 30–44 — moderate-to-severe decrease
- G4: eGFR 15–29 — severely decreased (pre-dialysis planning)
- G5: eGFR <15 — kidney failure (dialysis or transplant)
Albuminuria categories: A1 (<30 mg/g, normal), A2 (30–300 mg/g, moderately increased = "microalbuminuria"), A3 (>300 mg/g, severely increased = "macroalbuminuria"). Higher G + higher A = exponentially higher risk.
eGFR Equations — From Cockcroft-Gault to CKD-EPI 2021
The history of GFR estimation reflects evolving understanding of equity and accuracy:
- Cockcroft-Gault (1976): Estimates creatinine clearance using weight — overestimates GFR, not adjusted for body surface area, still used for drug dosing
- MDRD (1999): First equation to estimate GFR directly from serum creatinine + age + sex + race. Inaccurate at higher GFR (>60)
- CKD-EPI (2009): More accurate across all GFR ranges, but included a race coefficient that was debated
- CKD-EPI (2021): Removed race variable entirely — endorsed by NKF and ASN task force. Uses creatinine alone, or creatinine + cystatin C for improved accuracy. This change acknowledged that race is a social construct and the coefficient was based on biased reference populations
Key insight: The creatinine-cystatin C combined equation (CKD-EPI 2021) is the most accurate eGFR estimate available, especially in populations where creatinine alone may be misleading (extremes of muscle mass, vegetarians, amputees).
import numpy as np
import matplotlib.pyplot as plt
# CKD staging distribution in a hypothetical clinic population
stages = ['G1\n≥90', 'G2\n60-89', 'G3a\n45-59', 'G3b\n30-44', 'G4\n15-29', 'G5\n<15']
patients = [120, 250, 180, 95, 45, 20]
colors = ['#2ecc71', '#27ae60', '#f39c12', '#e67e22', '#e74c3c', '#c0392b']
fig, ax = plt.subplots(figsize=(10, 6))
bars = ax.bar(stages, patients, color=colors, edgecolor='white', linewidth=1.5)
for bar, count in zip(bars, patients):
ax.text(bar.get_x() + bar.get_width()/2, bar.get_height() + 5,
str(count), ha='center', fontweight='bold', fontsize=12)
ax.set_xlabel('CKD Stage (eGFR mL/min/1.73m²)', fontsize=12)
ax.set_ylabel('Number of Patients', fontsize=12)
ax.set_title('CKD Stage Distribution — Nephrology Clinic Cohort', fontsize=14, fontweight='bold')
ax.spines['top'].set_visible(False)
ax.spines['right'].set_visible(False)
plt.tight_layout()
plt.show()
Practice Exercises
Exercise 1: A patient presents with polyuria (10 L/day), dilute urine (osmolality 80 mOsm/kg), and hypernatraemia (Na⁺ 152 mmol/L). Administration of desmopressin has no effect on urine concentration. Diagnose and explain the biochemical defect.
Diagnosis: Nephrogenic diabetes insipidus
Key reasoning: The triad of massive dilute polyuria + hypernatraemia indicates ADH pathway failure. Desmopressin (synthetic ADH analogue) had no effect, ruling out central DI (where ADH production is deficient). In nephrogenic DI, the collecting duct is resistant to ADH — AQP2 water channels cannot be inserted into the apical membrane despite adequate V2 receptor stimulation. Common causes include lithium therapy (inhibits AQP2 expression via GSK3β), hypercalcaemia, and congenital V2 receptor mutations. Treatment: thiazide diuretics + amiloride (paradoxically reduce polyuria by enhancing proximal Na⁺ and water reabsorption), low-sodium diet, and NSAIDs (reduce prostaglandin-mediated antagonism of ADH).
Exercise 2: A 65-year-old diabetic patient has serum creatinine 150 µmol/L and urine ACR of 45 mg/mmol. Calculate the CKD stage and explain clinical implications.
Answer: Serum creatinine of 150 µmol/L corresponds to an eGFR of approximately 35–40 mL/min/1.73 m² (exact value depends on age/sex via CKD-EPI 2021 equation), placing the patient at CKD stage G3b (30–44). The urine ACR of 45 mg/mmol is in the A3 category (severely increased albuminuria, >30 mg/mmol). Combined G3b-A3 classification carries very high risk of progression to ESKD and cardiovascular events. Clinical implications: (1) Maximise RAAS blockade (ACEi/ARB to reduce proteinuria and slow progression — target ACR reduction >30%), (2) Add SGLT2 inhibitor (renoprotective independent of diabetes control), (3) BP target <130/80, (4) Avoid nephrotoxins (NSAIDs, contrast dye without pre-hydration), (5) Monitor potassium (combined ACEi + SGLT2i → hyperkalaemia risk), (6) Prepare for RRT planning if progression continues.
Exercise 3: Explain how the kidney compensates for a patient with chronic respiratory acidosis (COPD). Include the specific biochemical mechanisms and timeline.
Answer: In chronic respiratory acidosis (↑ CO₂ retention due to COPD), the kidney compensates by increasing H⁺ excretion and HCO₃⁻ generation. Timeline: (1) Acute (minutes to hours): Chemical buffers (haemoglobin, phosphate, proteins) absorb excess H⁺ — limited capacity. (2) Renal compensation (3–5 days to reach maximum): PCT increases NHE3 activity (more H⁺ secretion = more HCO₃⁻ reclamation); glutaminase activity upregulates (more NH₄⁺ production = more new HCO₃⁻ generation — can increase from 40 to 300 mmol/day); intercalated cells increase H⁺-ATPase and H⁺/K⁺-ATPase activity. Net result: plasma HCO₃⁻ rises (expected compensation: 3.5 mmol/L per 10 mmHg ↑ in PaCO₂), which partially restores the HCO₃⁻:CO₂ ratio toward 20:1, moving pH back toward normal but never fully correcting it. ABG pattern: pH slightly low (7.32–7.35), ↑PaCO₂, significantly ↑HCO₃⁻ (distinguishes chronic from acute).
Exercise 4: A patient on an ACE inhibitor presents with K⁺ of 6.2 mmol/L. Explain the biochemical mechanism and immediate management priorities.
Answer: ACE inhibitors reduce angiotensin II formation → reduced aldosterone secretion. Since aldosterone stimulates ENaC (Na⁺ reabsorption) and ROMK (K⁺ secretion) in the collecting duct, hypoaldosteronism means less K⁺ is secreted into the tubular lumen → hyperkalaemia. At K⁺ 6.2 mmol/L, this is a medical emergency. Immediate management: (1) Cardiac protection: IV calcium gluconate 10% (stabilises myocardial membrane — does NOT lower K⁺, works in 1–3 minutes, lasts 30–60 min); (2) Shift K⁺ intracellularly: Insulin 10 units + 50 mL 50% glucose IV (stimulates Na⁺/K⁺-ATPase → K⁺ enters cells, onset 15–30 min), nebulised salbutamol (β₂-agonist → ↑Na⁺/K⁺-ATPase), sodium bicarbonate if acidotic; (3) Remove K⁺ from body: Calcium resonium/patiromer/SZC (potassium binders, oral/rectal), loop diuretics (if adequate renal function), haemodialysis (if refractory or severe AKI); (4) Stop offending drug — withhold ACEi, review other K⁺-raising medications (spironolactone, amiloride, trimethoprim). Obtain an ECG immediately — look for peaked T waves, widened QRS, sine wave pattern.
Exercise 5: Compare and contrast the mechanisms of action of thiazide diuretics, loop diuretics, and potassium-sparing diuretics at the nephron level.
Answer: (1) Loop diuretics (furosemide, bumetanide): Block NKCC2 transporter in the thick ascending limb of the Loop of Henle → inhibit reabsorption of Na⁺, K⁺, and 2Cl⁻. This is the most powerful diuretic class because 25% of filtered Na⁺ is reabsorbed here. Also disrupts the medullary concentration gradient (less NaCl deposited in interstitium → impaired concentrating ability). Side effects: hypokalaemia (more Na⁺ delivered distally → more K⁺ secreted via ROMK), hypocalcaemia (loss of lumen-positive potential → less paracellular Ca²⁺ reabsorption), metabolic alkalosis, ototoxicity. (2) Thiazide diuretics (hydrochlorothiazide, bendroflumethiazide): Block NCC (Na⁺/Cl⁻ co-transporter) in the DCT — only ~5% of Na⁺ reabsorbed here, so weaker diuretic effect. However, by blocking NCC, more Ca²⁺ is reabsorbed (upregulated TRPV5/calbindin pathway) → used in calcium nephrolithiasis. Side effects: hypokalaemia, hypercalcaemia, hyperglycaemia, hyperuricaemia. (3) K⁺-sparing diuretics: Two subtypes — (a) Aldosterone antagonists (spironolactone, eplerenone): Block mineralocorticoid receptor → reduce ENaC and ROMK expression → less Na⁺ reabsortion, less K⁺ secretion; (b) ENaC blockers (amiloride, triamterene): Directly block the epithelial sodium channel. Both are weak diuretics alone but critically prevent hypokalaemia when combined with loop or thiazide diuretics.
Kidney Biochemistry Worksheet
Use this interactive worksheet to consolidate your understanding of renal biochemistry, acid-base balance, and clinical applications. Download your completed analysis as a Word, Excel, or PDF document.
Kidney Biochemistry Study Sheet
Complete the fields below to create a personalised kidney biochemistry summary. Download as Word, Excel, or PDF.
Conclusion & Next Steps
The kidneys are biochemical powerhouses that filter 180 litres of plasma daily, reclaiming >99% of filtered water, sodium, and bicarbonate while precisely adjusting the excretion of every electrolyte and acid-base equivalent. From the glomerular filtration barrier that discriminates by size and charge, through the serial transport mechanisms of each nephron segment, to the hormonal integration of ADH, aldosterone, and ANP — renal biochemistry exemplifies how molecular mechanisms translate into whole-body homeostasis.
The clinical relevance is immediate: understanding SGLT2 inhibitor pharmacology, diuretic mechanisms, acid-base compensation patterns, and biomarker interpretation are daily requirements in modern medicine. As we move forward, this renal foundation will prove essential for understanding endocrine regulation in Part 14, where hormones that command the kidney — and many other organs — are explored in biochemical depth.