Physiology Mastery
Homeostasis & Feedback
Set points, feedback loops, allostasisNeurophysiology & Action Potentials
Neurons, action potentials, synapsesCardiac Electrophysiology & Hemodynamics
Heart rhythm, hemodynamics, cardiac outputRespiratory Mechanics & Gas Exchange
Breathing mechanics, gas exchange, V/QRenal Physiology & Fluid Balance
Nephron function, filtration, acid-baseGI Physiology & Absorption
Motility, secretion, nutrient absorptionEndocrine Regulation & Metabolism
Hormones, thyroid, adrenal, metabolismExercise Physiology & Adaptation
Acute responses, training adaptationsCellular & Membrane Physiology
Ion transport, signaling, second messengersBlood & Immune Physiology
Hematopoiesis, coagulation, immunityReproductive & Developmental
Reproduction, pregnancy, fetal physiologyIntegrative & Clinical Physiology
Stress, shock, sepsis, agingMulti-System Stress Responses
The stress response is the body's most dramatic demonstration of integrative physiology — a coordinated, multi-system reaction designed to maintain survival in the face of threat. What began as an evolutionary adaptation to predators now activates in response to examinations, traffic jams, and public speaking. Understanding the physiological cascades involved connects nearly every organ system covered in this series.
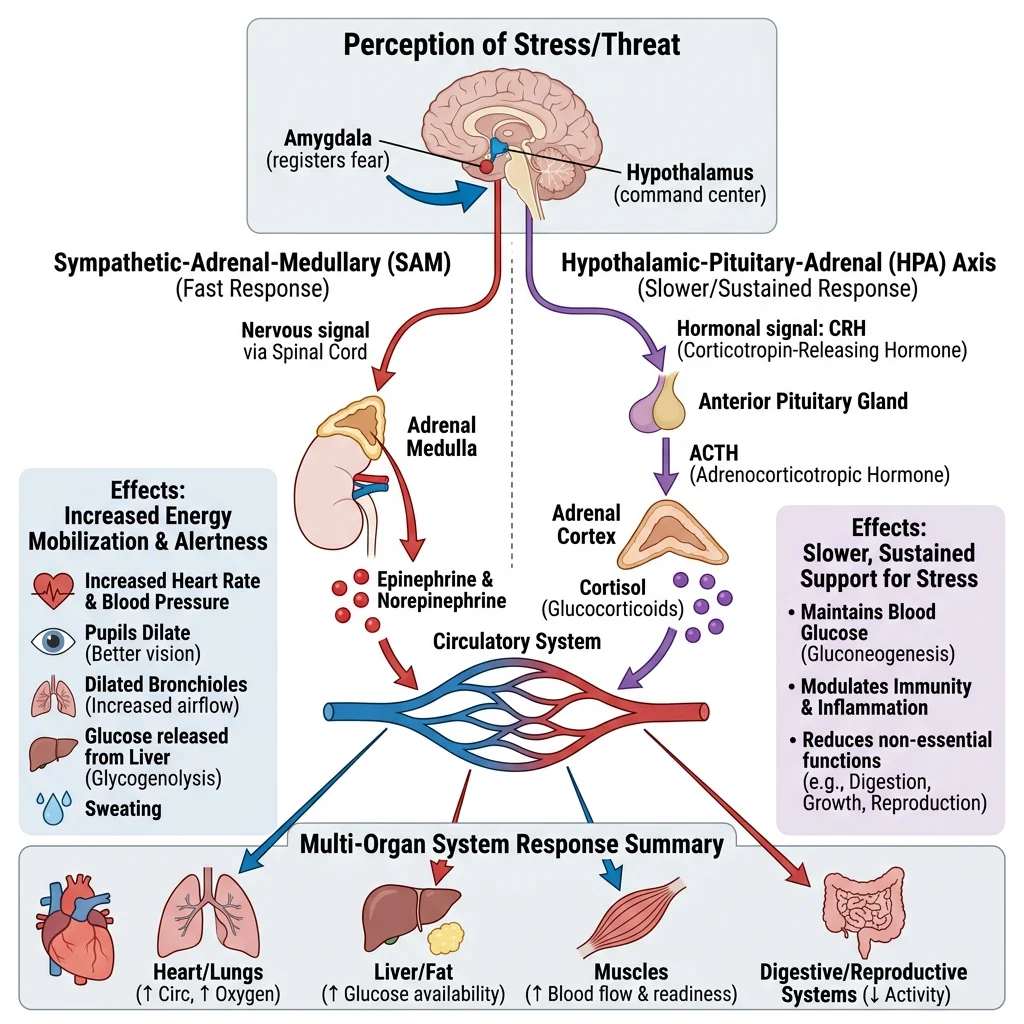
Fight-or-Flight & Sympathoadrenal Response
The sympathoadrenal system provides the immediate response to acute stress (seconds to minutes). Two parallel pathways activate simultaneously:
- Sympathetic nervous system: Preganglionic neurons (T1–L2) → postganglionic noradrenaline (norepinephrine) release at target organs: ↑ heart rate and contractility (β1), vasoconstriction of splanchnic and cutaneous beds (α1), bronchodilation (β2), mydriasis, ↓ GI motility, glycogenolysis in liver (β2)
- Adrenal medulla: Modified postganglionic sympathetic neurons secrete adrenaline (80%) and noradrenaline (20%) directly into the bloodstream → systemic hormonal amplification of the neural signal. Adrenaline's β2 effects (coronary/skeletal muscle vasodilation, bronchodilation) complement noradrenaline's α1 effects, creating a haemodynamic profile that redirects blood to muscles and the heart.
| System | Sympathoadrenal Effect | Receptor | Physiological Purpose |
|---|---|---|---|
| Heart | ↑ HR, ↑ contractility, ↑ conduction velocity | β1 | ↑ Cardiac output for fight/flight |
| Blood vessels | Vasoconstriction (skin, GI, renal); vasodilation (skeletal muscle, coronary) | α1 (constrict); β2 (dilate) | Redirect blood to muscles and brain |
| Lungs | Bronchodilation | β2 | ↑ Airflow for O₂ delivery |
| Liver | Glycogenolysis, gluconeogenesis | β2, α1 | ↑ Blood glucose (fuel for muscles/brain) |
| Eyes | Mydriasis (pupil dilation) | α1 | ↑ Visual acuity, wider field of vision |
| Adipose tissue | Lipolysis → FFA release | β3 | Alternative fuel substrate |
HPA Axis & Cortisol
While the sympathoadrenal system provides the immediate response, the hypothalamic-pituitary-adrenal (HPA) axis mediates the sustained stress response (minutes to hours to days):
- Hypothalamic CRH: Stress signals (from amygdala, brainstem, cytokines) → paraventricular nucleus (PVN) → corticotropin-releasing hormone (CRH) + AVP release into hypophyseal portal blood
- Pituitary ACTH: CRH binds CRH-R1 on anterior pituitary corticotrophs → ACTH (adrenocorticotrophic hormone) release from POMC cleavage (β-endorphin co-released → stress-induced analgesia)
- Adrenal cortisol: ACTH stimulates zona fasciculata → cortisol synthesis (from cholesterol via desmolase/CYP11A1). Cortisol has a circadian rhythm (peak: 6–8 AM; nadir: midnight) that is overridden during stress.
Cortisol Actions — The "Survival Hormone"
- Metabolic: ↑ Gluconeogenesis (liver), ↑ proteolysis (muscle → amino acids for gluconeogenesis), ↑ lipolysis → ensuring blood glucose supply ("permissive effect" on glucagon and adrenaline)
- Anti-inflammatory: ↓ NF-κB → ↓ IL-1, IL-6, TNF-α; ↓ prostaglandin synthesis (via lipocortin/annexin-1 → ↓ PLA₂); ↓ leukocyte migration. This is why corticosteroids are powerful anti-inflammatory drugs.
- Immunosuppressive: ↓ T-cell proliferation, ↓ antibody production — protective against autoimmune damage but increases infection susceptibility with chronic elevation
- Cardiovascular: Permissive effect on catecholamines (maintains vascular responsiveness to noradrenaline — addisonian crisis involves cardiovascular collapse because cortisol deficiency → impaired vasoconstrictor response)
- CNS: Enhances alertness and memory consolidation acutely; chronic excess → hippocampal atrophy, depression, cognitive impairment
Metabolic Stress Response
Major physiological stress (surgery, trauma, sepsis, burns) triggers a profound hypermetabolic, catabolic state orchestrated by cortisol, catecholamines, glucagon, and cytokines:
- "Stress hyperglycaemia": Cortisol + adrenaline + glucagon → hepatic gluconeogenesis + glycogenolysis + peripheral insulin resistance → blood glucose can exceed 200 mg/dL even in non-diabetics. The teleological purpose: ensure glucose delivery to the brain and immune cells.
- Protein catabolism: Cortisol drives skeletal muscle proteolysis → amino acids (especially alanine, glutamine) fuel hepatic gluconeogenesis and serve as substrates for acute phase protein synthesis (CRP, fibrinogen, ferritin). Severe catabolic states can cause loss of 0.5–1 kg lean body mass per day.
- Lipolysis: Catecholamines activate hormone-sensitive lipase → free fatty acids and glycerol → FFA oxidation provides energy for non-essential tissues, sparing glucose for the brain.
- Acute phase response: IL-6 (from macrophages) → liver → ↑ CRP, fibrinogen, haptoglobin, ferritin, complement, α1-antitrypsin; ↓ albumin, transferrin (negative acute phase proteins). CRP rises 1000-fold and is the most widely used clinical marker of systemic inflammation.
Immune-Neuroendocrine Crosstalk
The nervous, endocrine, and immune systems communicate bidirectionally — a concept known as psychoneuroimmunology:
- Brain → Immune: Sympathetic innervation of lymphoid organs (spleen, lymph nodes, thymus); noradrenaline modulates immune cell function via β2 receptors. Cortisol redistributes lymphocytes and modulates cytokine production.
- Immune → Brain: Pro-inflammatory cytokines (IL-1, IL-6, TNF-α) signal to the brain via: (1) circumventricular organs (no BBB); (2) vagal afferents from the periphery; (3) active transport across the BBB. This induces "sickness behaviour" — fever, fatigue, anorexia, social withdrawal, hyperalgesia — an adaptive conservational strategy that redirects energy to fighting infection.
- The Vagal Anti-Inflammatory Reflex: Kevin Tracey's discovery (2000) that vagus nerve stimulation suppresses TNF-α production via the "cholinergic anti-inflammatory pathway": vagal efferents → splenic nerve → T-cells release ACh → macrophage α7 nAChR → ↓ NF-κB → ↓ TNF-α. This is being exploited as a therapeutic target — vagus nerve stimulation devices for rheumatoid arthritis and inflammatory bowel disease.
Shock States
Shock is defined as inadequate tissue perfusion and cellular oxygen delivery relative to metabolic demand, resulting in cellular hypoxia and — if untreated — irreversible organ damage and death. Shock is not a blood pressure number; it is a physiological state that reflects failure of the most fundamental job of the cardiovascular system: delivering oxygen to cells.
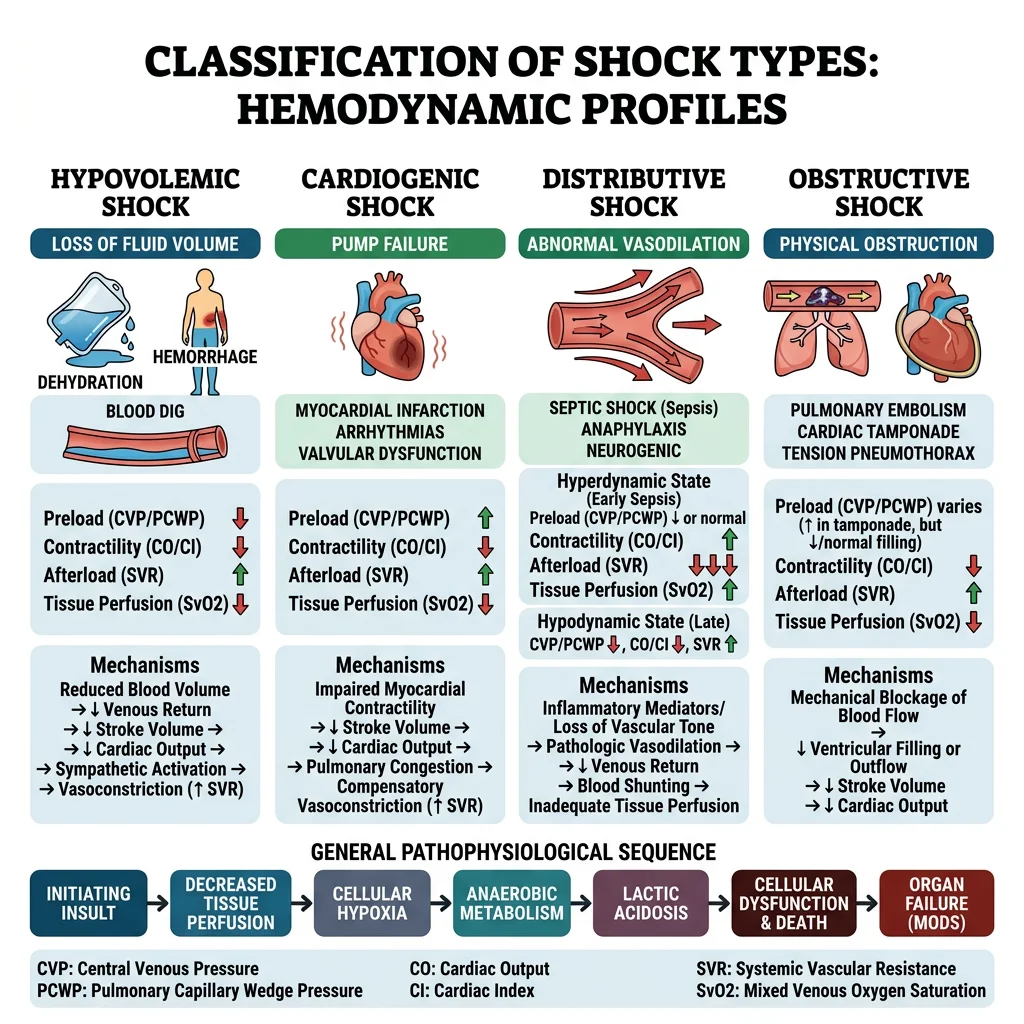
Hypovolemic Shock
Cause: ↓ Circulating volume — haemorrhage (most common), dehydration, burns (third-spacing), pancreatitis. Haemorrhagic shock is classified by volume lost:
| Class | Blood Loss | HR | BP | RR | Mental Status | Urine Output |
|---|---|---|---|---|---|---|
| I | <750 mL (<15%) | <100 | Normal | 14–20 | Slightly anxious | >30 mL/h |
| II | 750–1500 mL (15-30%) | 100–120 | Normal (narrowed pulse pressure) | 20–30 | Anxious | 20–30 mL/h |
| III | 1500–2000 mL (30-40%) | >120 | ↓↓ | 30–40 | Confused | 5–15 mL/h |
| IV | >2000 mL (>40%) | >140 or bradycardia | Profoundly ↓↓↓ | >35 | Lethargic/obtunded | Negligible |
Pathophysiology: ↓ Preload → ↓ SV → ↓ CO → baroreceptor activation → ↑ sympathetic tone → tachycardia, vasoconstriction (cold, clammy skin), ↑ contractility. RAAS activation → Na⁺/H₂O retention. ADH release → water retention. These compensatory mechanisms maintain BP initially ("compensated shock" — note BP is the last parameter to fall).
Cardiogenic Shock
Cause: Pump failure — massive MI (>40% of LV myocardium), acute mitral regurgitation, VSD, myocarditis, end-stage cardiomyopathy.
Pathophysiology: ↓ Contractility → ↓ SV → ↓ CO despite adequate/increased preload. Blood backs up → ↑ LV end-diastolic pressure → ↑ LA pressure → pulmonary oedema (distinguishing feature). The failing ventricle enters a vicious cycle: ↓ CO → ↓ coronary perfusion → further ischaemia → further ↓ contractility.
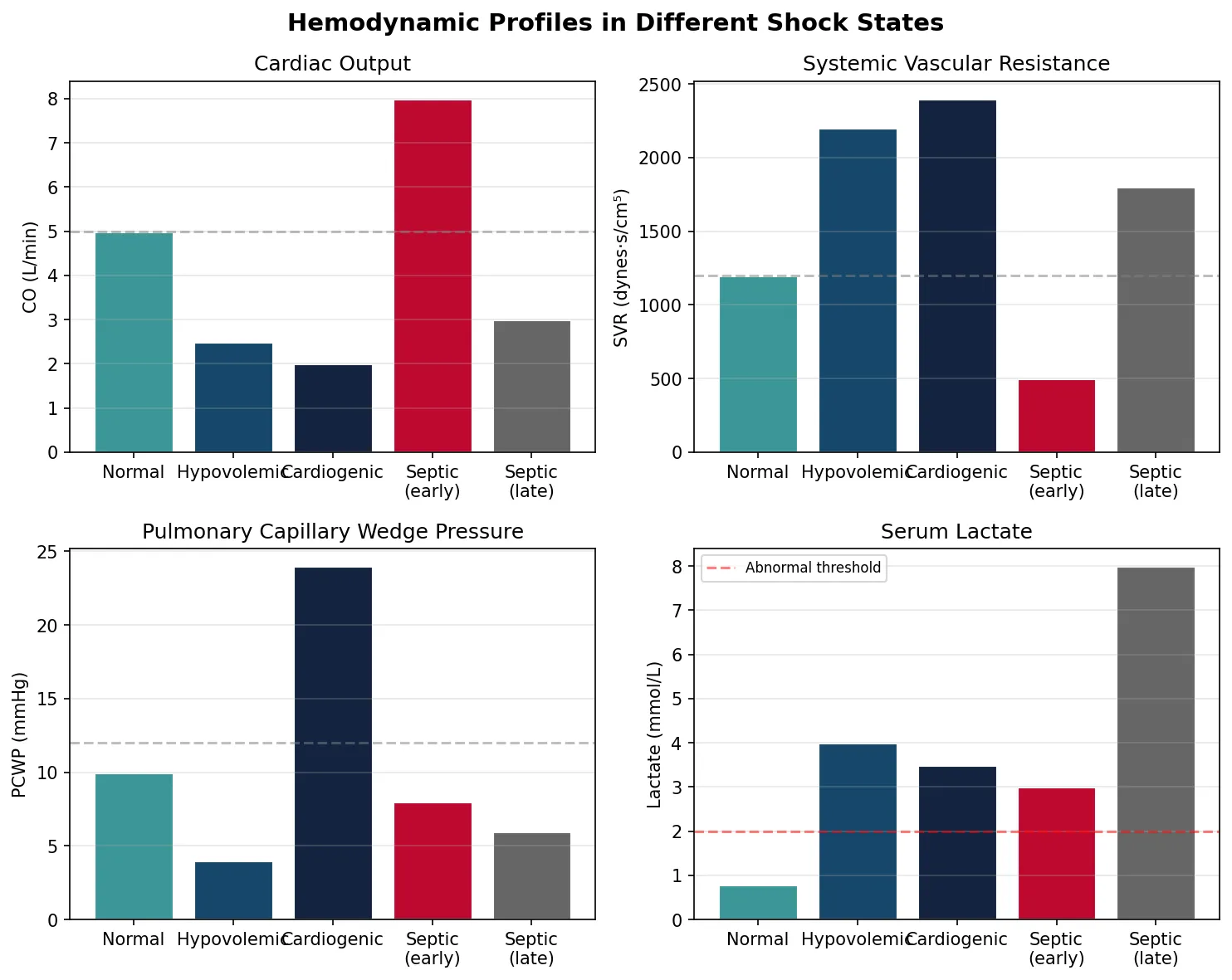
Haemodynamic profile (Swan-Ganz): ↑ PCWP (>18 mmHg), ↓ CI (<2.2 L/min/m²), ↑ SVR (compensatory vasoconstriction). Compare with hypovolemic shock: ↓ PCWP, ↓ CI, ↑ SVR.
Distributive Shock
The hallmark of distributive shock is pathological vasodilation — the "container" (vascular system) becomes too large for the volume, despite normal or even increased cardiac output. Three main types:
- Septic shock: Most common and lethal. Infection → systemic inflammation → NO and prostacyclin → massive vasodilation + capillary leak + myocardial depression. Warm shock (early) → hyperdynamic: ↑ CO, ↓ SVR, warm extremities. Cold shock (late/decompensated) → ↓ CO, vasoconstriction.
- Anaphylactic shock: IgE-mediated mast cell degranulation → histamine, tryptase, prostaglandins, leukotrienes → vasodilation, bronchospasm, angioedema. Onset within minutes of allergen exposure. Treatment: intramuscular adrenaline (anterolateral thigh, 0.5 mg of 1:1000) — the single most important intervention.
- Neurogenic shock: Spinal cord injury above T6 → loss of sympathetic tone → unopposed vagal activity → vasodilation + bradycardia (unique: other shock types cause tachycardia). "Warm, dry, bradycardic" patient.
Compensatory Mechanisms & Decompensation
The body's compensatory responses to shock integrate virtually every system we've studied:
| Mechanism | Mediator | Timing | Effect |
|---|---|---|---|
| Baroreceptor reflex | Carotid sinus, aortic arch → SNS | Seconds | ↑ HR, ↑ contractility, vasoconstriction |
| Chemoreceptor activation | Carotid/aortic bodies (↓ PaO₂, ↑ PaCO₂, ↓ pH) | Seconds-minutes | ↑ RR and depth → ↑ O₂ delivery, ↓ CO₂ |
| RAAS | Renin → angiotensin II → aldosterone | Minutes-hours | Vasoconstriction (AT-II); Na⁺/H₂O retention (aldosterone) |
| ADH (vasopressin) | Posterior pituitary | Minutes-hours | Water retention (V2 renal); vasoconstriction (V1 vascular) |
| Transcapillary refill | Starling forces | Minutes-hours | ↓ Capillary hydrostatic pressure → interstitial fluid moves into vascular space (up to 1 L in first hour) |
| Erythropoietin | Kidney (peritubular fibroblasts) | Days | ↑ RBC production → restores O₂ carrying capacity |
Decompensation occurs when compensatory mechanisms are overwhelmed or become harmful: sustained vasoconstriction → gut ischaemia → bacterial translocation → sepsis ("gut as the motor of MOF"); myocardial depression from acidosis and cytokines; DIC from coagulopathy; renal failure from prolonged hypoperfusion. This progression to multi-organ failure (MOF) is the terminal common pathway of untreated shock.
Haemorrhagic Shock — Trauma Scenario
A 28-year-old male arrives at the ED after a motor vehicle accident. GCS 13, HR 128, BP 85/60, RR 32, SpO₂ 94% on room air, cool/clammy extremities, distended abdomen. Two large-bore IVs established. FAST ultrasound positive for free fluid in Morrison's pouch and the pelvis.
Physiological Analysis: Class III haemorrhagic shock (~1.5–2 L blood loss). Tachycardia and vasoconstriction (baroreceptor reflex); low BP indicates decompensation; tachypnoea (chemoreceptor stimulation from metabolic acidosis); confusion (cerebral hypoperfusion); oliguria (renal vasoconstriction + ADH). The FAST-positive abdomen suggests intra-abdominal haemorrhage likely requiring emergent laparotomy.
Management priorities: (1) Activate massive transfusion protocol (1:1:1 ratio of packed RBCs, FFP, platelets); (2) permissive hypotension (target SBP ~80–90 mmHg until surgical haemostasis — aggressive fluid resuscitation before bleeding is controlled worsens coagulopathy by dilution and hypothermia); (3) emergent surgical intervention; (4) tranexamic acid (within 3 hours — CRASH-2 trial showed ↓ mortality); (5) warm patient (hypothermia → coagulopathy → acidosis: the "lethal triad").
Sepsis Physiology
Sepsis is the leading cause of death in hospitalised patients and represents the most complex multi-system derangement in clinical physiology. The modern definition (Sepsis-3, 2016) defines sepsis as "life-threatening organ dysfunction caused by a dysregulated host response to infection" — emphasising that it is the host's response, not the infection itself, that causes the damage.
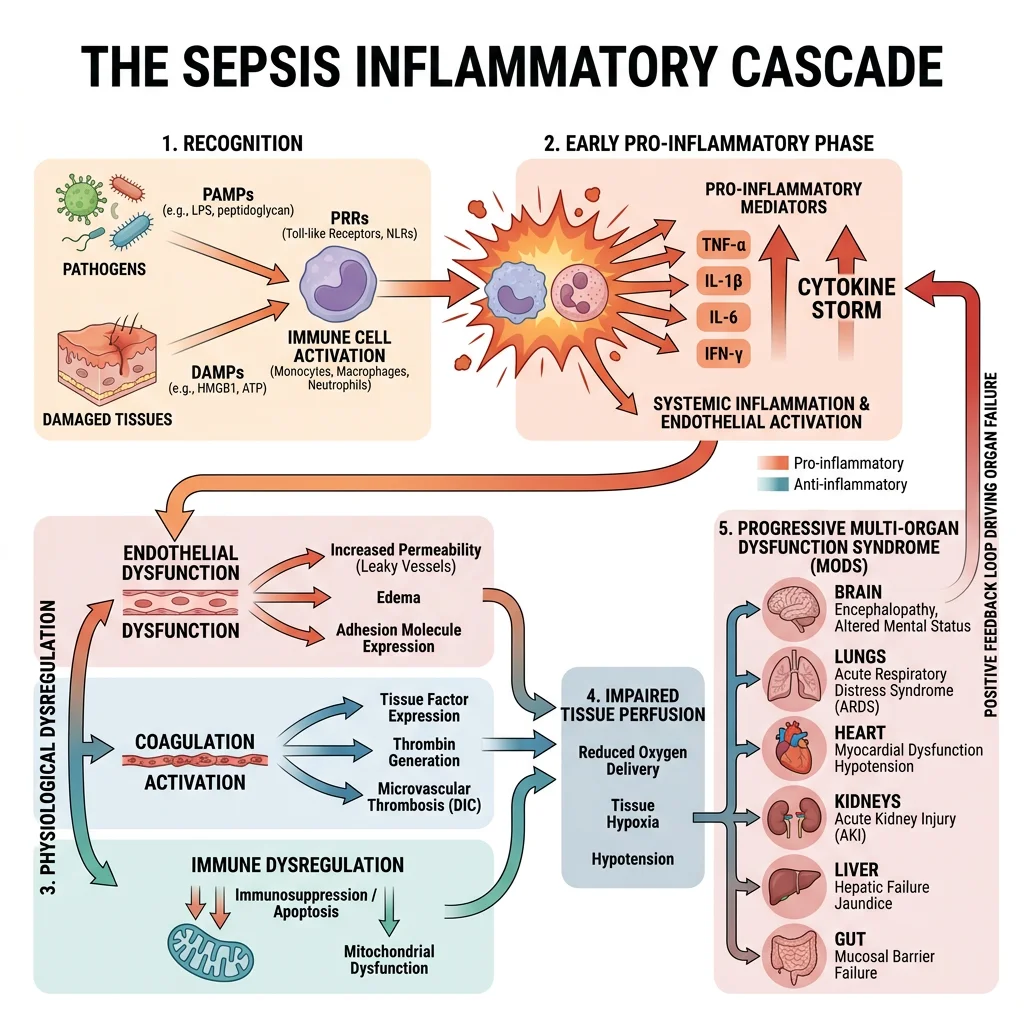
Systemic Inflammatory Response
The sepsis cascade begins when the innate immune system detects pathogen-associated molecular patterns (PAMPs) — lipopolysaccharide (LPS) from Gram-negative bacteria is the prototype. PAMPs bind Toll-like receptors (TLRs) on macrophages and dendritic cells → NF-κB activation → massive release of pro-inflammatory cytokines:
- TNF-α: Released within minutes; activates endothelium (↑ adhesion molecules), activates coagulation, induces fever, promotes apoptosis. The earliest and most potent mediator.
- IL-1β: Synergises with TNF-α; pyrogenic; stimulates hepatic acute-phase proteins.
- IL-6: Major driver of CRP synthesis; correlates with sepsis severity. The most reliable clinical biomarker of systemic inflammation.
- IL-8: Neutrophil chemotaxis → recruits neutrophils to sites of infection (and unfortunately, also to healthy tissues in sepsis).
Simultaneously, damage-associated molecular patterns (DAMPs) — released from injured host cells (HMGB1, mitochondrial DNA, ATP) — amplify the response through the same TLR pathways, creating a positive feedback loop that can become self-sustaining even after the infection is controlled.
Cytokine Storm & Organ Damage
The "cytokine storm" describes the uncontrolled, systemic release of pro-inflammatory cytokines causing collateral damage to host tissues. Each organ system suffers in different ways:
- Lungs — ARDS: Neutrophil infiltration → endothelial damage → capillary leak → non-cardiogenic pulmonary oedema → diffuse alveolar damage. Compliance drops, V/Q mismatch worsens → refractory hypoxaemia (PaO₂/FiO₂ < 300 = ALI; < 200 = ARDS).
- Kidneys — Acute Kidney Injury: Combination of tubular injury (ischaemia + nephrotoxins), microvascular dysfunction, and inflammation → oliguria → uraemia → metabolic acidosis.
- Liver — Shock liver: Hypoperfusion + inflammatory injury → massive transaminase elevation ("ischaemic hepatitis"); impaired conjugation, synthetic function, and clearance.
- Brain — Septic encephalopathy: BBB disruption, cerebral oedema, microglial activation → delirium, confusion, coma. Present in up to 70% of sepsis patients. Independent predictor of mortality.
- Heart — Septic cardiomyopathy: Cytokines (TNF-α, IL-1) + NO → myocardial depression. EF may drop from 60% to 20-30%. Remarkably, this is usually reversible in survivors.
Hemodynamic Derangements
Septic shock creates a unique and paradoxical haemodynamic state:
- Massive vasodilation: iNOS upregulation → excessive nitric oxide → systemic vasodilation → ↓↓ SVR (the defining feature)
- Capillary leak: Endothelial glycocalyx degradation → plasma extravasation → interstitial oedema → relative hypovolaemia
- Hyperdynamic early phase: ↑ CO (compensatory) despite vasodilation — "warm shock" (flushed, warm skin). This distinguishes septic from cardiogenic shock.
- Microcirculatory dysfunction: Despite adequate macrocirculatory parameters (MAP > 65), microvascular shunting, thrombosis, and heterogeneous flow → oxygen delivery fails at the tissue level. This explains why patients can die of organ failure despite achieving haemodynamic targets.
- Lactate elevation: Traditionally attributed to tissue hypoxia → anaerobic glycolysis. Now recognised to also reflect aerobic glycolysis driven by catecholamines (↑ Na⁺/K⁺-ATPase activity) and impaired hepatic lactate clearance. Serum lactate > 2 mmol/L with persistent hypotension after fluid resuscitation defines septic shock (Sepsis-3).
import numpy as np
import matplotlib.pyplot as plt
# Simulate shock hemodynamic profiles
shock_types = ['Normal', 'Hypovolemic', 'Cardiogenic', 'Septic\n(early)', 'Septic\n(late)']
co = [5.0, 2.5, 2.0, 8.0, 3.0] # Cardiac Output (L/min)
svr = [1200, 2200, 2400, 500, 1800] # SVR (dynes·s/cm⁵)
pcwp = [10, 4, 24, 8, 6] # PCWP (mmHg)
lactate = [0.8, 4.0, 3.5, 3.0, 8.0] # Lactate (mmol/L)
fig, axes = plt.subplots(2, 2, figsize=(10, 8))
fig.suptitle('Hemodynamic Profiles in Different Shock States', fontsize=14, fontweight='bold')
colors = ['#3B9797', '#16476A', '#132440', '#BF092F', '#666666']
# CO
axes[0,0].bar(shock_types, co, color=colors, edgecolor='white', linewidth=1.5)
axes[0,0].set_ylabel('CO (L/min)')
axes[0,0].set_title('Cardiac Output')
axes[0,0].axhline(y=5.0, color='gray', linestyle='--', alpha=0.5, label='Normal')
axes[0,0].grid(axis='y', alpha=0.3)
# SVR
axes[0,1].bar(shock_types, svr, color=colors, edgecolor='white', linewidth=1.5)
axes[0,1].set_ylabel('SVR (dynes·s/cm⁵)')
axes[0,1].set_title('Systemic Vascular Resistance')
axes[0,1].axhline(y=1200, color='gray', linestyle='--', alpha=0.5)
axes[0,1].grid(axis='y', alpha=0.3)
# PCWP
axes[1,0].bar(shock_types, pcwp, color=colors, edgecolor='white', linewidth=1.5)
axes[1,0].set_ylabel('PCWP (mmHg)')
axes[1,0].set_title('Pulmonary Capillary Wedge Pressure')
axes[1,0].axhline(y=12, color='gray', linestyle='--', alpha=0.5)
axes[1,0].grid(axis='y', alpha=0.3)
# Lactate
axes[1,1].bar(shock_types, lactate, color=colors, edgecolor='white', linewidth=1.5)
axes[1,1].set_ylabel('Lactate (mmol/L)')
axes[1,1].set_title('Serum Lactate')
axes[1,1].axhline(y=2.0, color='red', linestyle='--', alpha=0.5, label='Abnormal threshold')
axes[1,1].legend(fontsize=8)
axes[1,1].grid(axis='y', alpha=0.3)
plt.tight_layout()
plt.show()
print("Key: Septic shock → ↓SVR, ↑CO (early), ↑lactate. Cardiogenic → ↓CO, ↑PCWP, ↑SVR.")
Sepsis-Induced Coagulopathy
Disseminated intravascular coagulation (DIC) is the prototypical sepsis coagulopathy — a paradox of simultaneous thrombosis and bleeding:
- Pro-coagulant activation: TNF-α + IL-1 → tissue factor expression on endothelium and monocytes → extrinsic pathway activation → widespread microthrombi in the microcirculation → organ ischaemia
- Anticoagulant suppression: ↓ Antithrombin III (consumed), ↓ protein C and S, ↓ thrombomodulin (endothelial damage)
- Fibrinolytic shutdown: ↑ PAI-1 (plasminogen activator inhibitor-1) → impaired clot breakdown → persistent microthrombi
- Consumption coagulopathy: Clotting factors and platelets consumed by widespread thrombus formation → ↑ PT/INR, ↑ APTT, ↓ platelets, ↓ fibrinogen, ↑ D-dimer, ↑ FDP → bleeding from IV sites, mucosal surfaces, surgical wounds
Septic Shock — Integrating All Systems
A 62-year-old woman with a UTI develops confusion, HR 115, BP 78/45, RR 28, temp 39.5°C, SpO₂ 91% on room air, lactate 5.2 mmol/L, WBC 22,000 with left shift, creatinine 2.8 mg/dL (baseline 0.9), platelets 85,000, INR 1.8. Blood cultures grow E. coli.
Multi-system analysis: Infection (UTI → E. coli bacteraemia) triggered SIRS → sepsis → septic shock (hypotension + lactate > 2). Cardiovascular: ↓ SVR (vasodilation), ↑ CO initially (hyperdynamic). Renal: AKI (Cr 2.8 from 0.9 — KDIGO stage 2). Haematological: DIC developing (↓ platelets, ↑ INR). CNS: septic encephalopathy (confusion). Respiratory: early ARDS (SpO₂ 91%, tachypnoea). Metabolic: lactic acidosis (anaerobic metabolism).
Management (Hour-1 Bundle): (1) Measure lactate; (2) blood cultures × 2 before antibiotics; (3) IV broad-spectrum antibiotics within 1 hour — the most time-critical intervention (every hour of delay ↑ mortality ~7.6%); (4) 30 mL/kg crystalloid bolus for hypotension or lactate ≥ 4; (5) vasopressors (noradrenaline first-line) if MAP < 65 mmHg after fluids → target MAP ≥ 65. If refractory → add vasopressin → then consider hydrocortisone 200 mg/day (relative adrenal insufficiency).
Thermoregulation Extremes
Core temperature is one of the most tightly regulated physiological variables (set point ~37°C ± 0.5°C), controlled by the hypothalamic thermoregulatory centre (preoptic/anterior hypothalamus). Temperature extremes — whether environmental or pathological — stress every organ system and serve as an excellent model for integrative physiology.
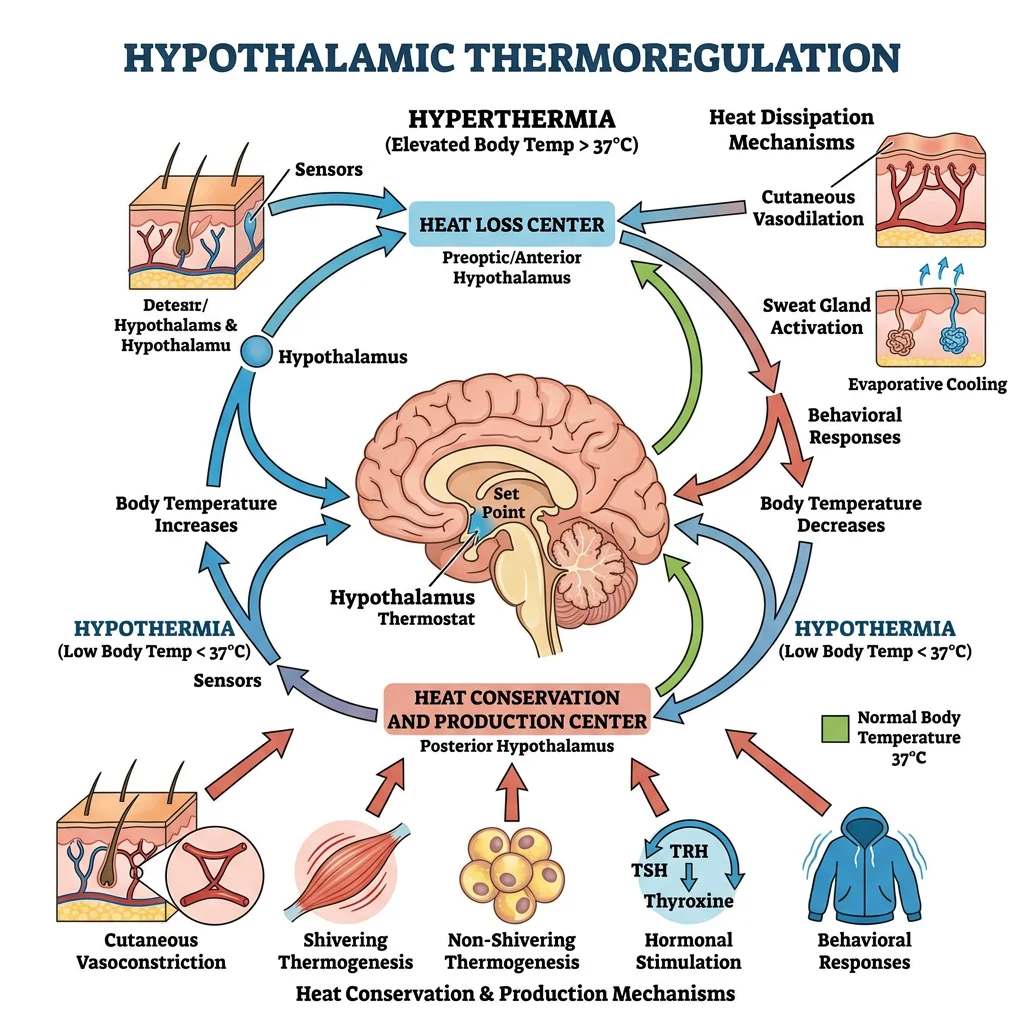
Hypothermia Physiology
Hypothermia (core temperature < 35°C) triggers a graded physiological response that eventually overwhelms compensatory mechanisms:
| Severity | Core Temp | Cardiovascular | Neurological | Metabolic | Other |
|---|---|---|---|---|---|
| Mild | 32–35°C | Tachycardia, vasoconstriction, ↑ BP | Confusion, dysarthria, poor judgement | ↑ Metabolic rate (shivering), ↑ catecholamines | Shivering maximal; "cold diuresis" (peripheral vasoconstriction → central volume shift → ↓ ADH) |
| Moderate | 28–32°C | Bradycardia, ↓ CO, atrial fibrillation, J (Osborn) waves on ECG | Stupor, loss of shivering (below ~30°C), amnesia | ↓ Metabolic rate; ↓ O₂ consumption 6% per 1°C drop | Paradoxical undressing; coagulopathy (enzyme dysfunction) |
| Severe | <28°C | VF risk highest at ~28°C; asystole below ~20°C | Coma, absent reflexes | Near-zero metabolic rate; cellular hibernation | "No one is dead until warm and dead" — rewarming may restore circulation |
Hyperthermia & Heat Stroke
Hyperthermia occurs when heat generation or absorption exceeds the body's capacity for dissipation. Unlike fever, the thermoregulatory set point is not elevated — the hypothalamus is trying to cool the body but cannot.
Heat stroke (core temp > 40°C with CNS dysfunction) is a medical emergency with mortality up to 50% if untreated. Pathophysiology: extreme heat → protein denaturation → direct cellular toxicity; splanchnic vasoconstriction (blood redirected to skin for cooling) → gut barrier failure → endotoxin translocation → SIRS; DIC from endothelial damage; rhabdomyolysis → myoglobinuria → AKI.
Fever Mechanisms
Fever is fundamentally different from hyperthermia — it involves an upward resetting of the hypothalamic set point by endogenous pyrogens:
- Infection → macrophages detect PAMPs → release endogenous pyrogens (IL-1, IL-6, TNF-α, IFN-α)
- Pyrogens reach the hypothalamus (via OVLT — organum vasculosum of the lamina terminalis, a circumventricular organ lacking BBB)
- Endothelial cells and perivascular glia produce PGE₂ (via COX-2 + mPGES-1)
- PGE₂ binds EP3 receptors on thermoregulatory neurons in the preoptic area → set point rises
- Body perceives current temperature as "too cold" → shivering, vasoconstriction, behavioural changes (seeking warmth) → temperature rises to the new set point
Antipyretics (paracetamol, NSAIDs) work by inhibiting COX → ↓ PGE₂ → set point returns to normal → sweating, vasodilation → temperature falls. They do not work in hyperthermia (set point is already normal).
Therapeutic Hypothermia
Targeted temperature management (TTM) exploits the neuroprotective effects of mild hypothermia (32–36°C) after cardiac arrest. The landmark HACA trial (2002) and Bernard et al. demonstrated improved neurological outcomes and survival in comatose post-cardiac arrest patients cooled to 32–34°C for 24 hours. The mechanism: ↓ metabolic demand → ↓ O₂ consumption → ↓ excitotoxic neurotransmitter release → ↓ free radical production → ↓ apoptosis. Current guidelines (TTM2 trial, 2021) suggest targeting normothermia (<37.5°C) may be equivalent to active cooling to 33°C, but fever prevention remains critical.
Aging Physiology
Ageing represents the gradual decline of physiological function across every organ system — reducing physiological reserve (the capacity to respond to stress) and increasing vulnerability to disease. While ageing is inevitable, understanding the physiological changes helps distinguish normal ageing from disease and identify targets for intervention.
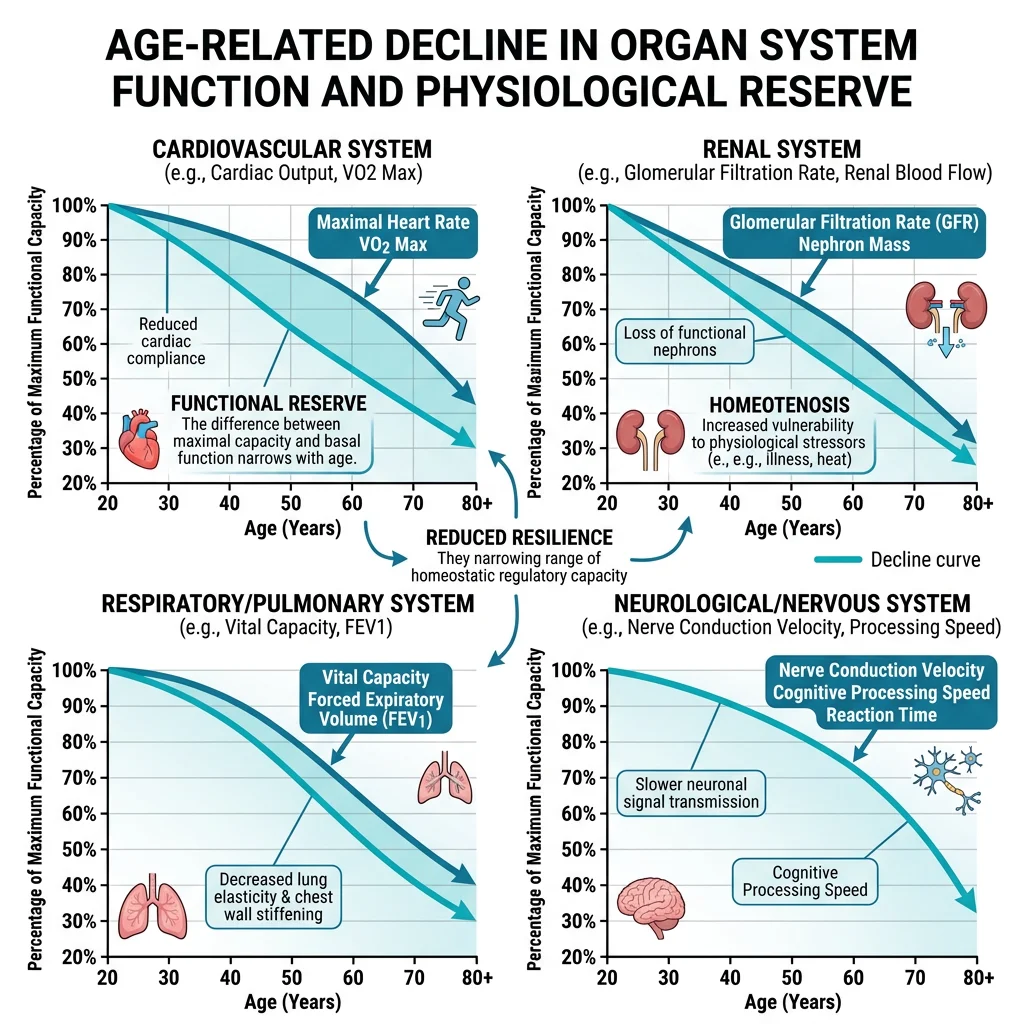
Cardiovascular Aging
- Arterial stiffening: ↑ Collagen, ↓ elastin, AGE (advanced glycation end-product) crosslinking → ↑ pulse wave velocity → ↑ systolic BP → isolated systolic hypertension (the most common form of hypertension in the elderly). Wide pulse pressure (SBP − DBP > 60 mmHg) is an independent cardiovascular risk factor.
- Diastolic dysfunction: LV wall thickening + ↓ compliance → impaired relaxation → heart fills adequately at rest but cannot increase preload during exercise → exercise intolerance (heart failure with preserved ejection fraction, HFpEF, is the most common form of heart failure in the elderly)
- Conduction system degeneration: Fibrosis of the SA node → ↓ maximum heart rate (~220 − age); AV node slowing → first-degree heart block; atrial fibrillation prevalence increases exponentially (from ~1% at 50 to ~10% at 80)
- Baroreceptor blunting: ↓ Sensitivity → impaired response to orthostatic stress → orthostatic hypotension (prevalence ~20% in those >65) → falls → hip fractures → a major cause of morbidity
Musculoskeletal Decline
- Sarcopenia: Age-related loss of skeletal muscle mass and strength — begins at ~30 years, accelerates after 60 (loss of ~3–8% of muscle mass per decade). Mechanism: ↓ anabolic hormones (testosterone, GH, IGF-1), ↓ motor neuron numbers (denervation of fast-twitch type II fibres), ↑ inflammatory cytokines (TNF-α, IL-6), reduced satellite cell activation. Sarcopenia → frailty → falls → disability → loss of independence.
- Osteoporosis: ↓ Bone mineral density (peak bone mass ~30 years; decline thereafter, accelerated in postmenopausal women due to oestrogen withdrawal → ↑ osteoclast activity > osteoblast activity). DEXA T-score ≤ −2.5 = osteoporosis. Fragility fractures (hip, vertebral, wrist) are the clinical consequence.
- Osteoarthritis: Cartilage degradation from cumulative mechanical stress + inflammatory mediators → pain, stiffness, reduced mobility. The most common joint disease worldwide.
Neurological Aging
- Brain atrophy: ~0.5% volume loss per year after 60; prefrontal cortex and hippocampus are particularly affected → ↓ executive function, ↓ working memory, ↓ processing speed. Importantly, crystallised intelligence (accumulated knowledge) is largely preserved.
- Neurotransmitter decline: ↓ Dopamine (substantia nigra) → Parkinson's risk; ↓ acetylcholine (nucleus basalis of Meynert) → Alzheimer's risk; ↓ serotonin → depression vulnerability
- Sleep architecture changes: ↓ Slow-wave sleep (stage N3), ↑ sleep fragmentation, earlier circadian phase (advanced sleep phase) → daytime somnolence, ↓ memory consolidation
- Peripheral neuropathy: ↓ Nerve conduction velocity (~1 m/s per decade), ↓ vibration sense, ↓ proprioception → contributes to falls
Metabolic & Endocrine Changes
- Insulin resistance: ↓ Muscle mass (major glucose disposal site) + ↑ visceral fat + ↓ physical activity → progressive insulin resistance → type 2 diabetes prevalence peaks at 65–74 years
- Thyroid: ↑ Prevalence of subclinical hypothyroidism (↑ TSH, normal T4) — affects up to 15% of women >60. Symptoms overlap with "normal ageing" (fatigue, cold intolerance, weight gain, cognitive slowing) → frequently missed.
- Adrenal: ↓ DHEA and DHEA-S (~95% decline from peak at age 25 to age 85) — the "adrenopause." Clinical significance debated; DHEA supplements not proven beneficial.
- GH/IGF-1: ↓ GH secretion (~14% per decade after 30) — "somatopause." Contributes to ↓ muscle mass, ↑ fat, ↓ bone density. GH replacement is not recommended due to side effects (insulin resistance, fluid retention, possible cancer risk).
- Renal: GFR declines ~1 mL/min/year after age 40 (loss of nephrons). Serum creatinine may remain "normal" because ↓ muscle mass → ↓ creatinine production masks the decline. Drug dosing must be adjusted for estimated GFR (CKD-EPI or Cockcroft-Gault).
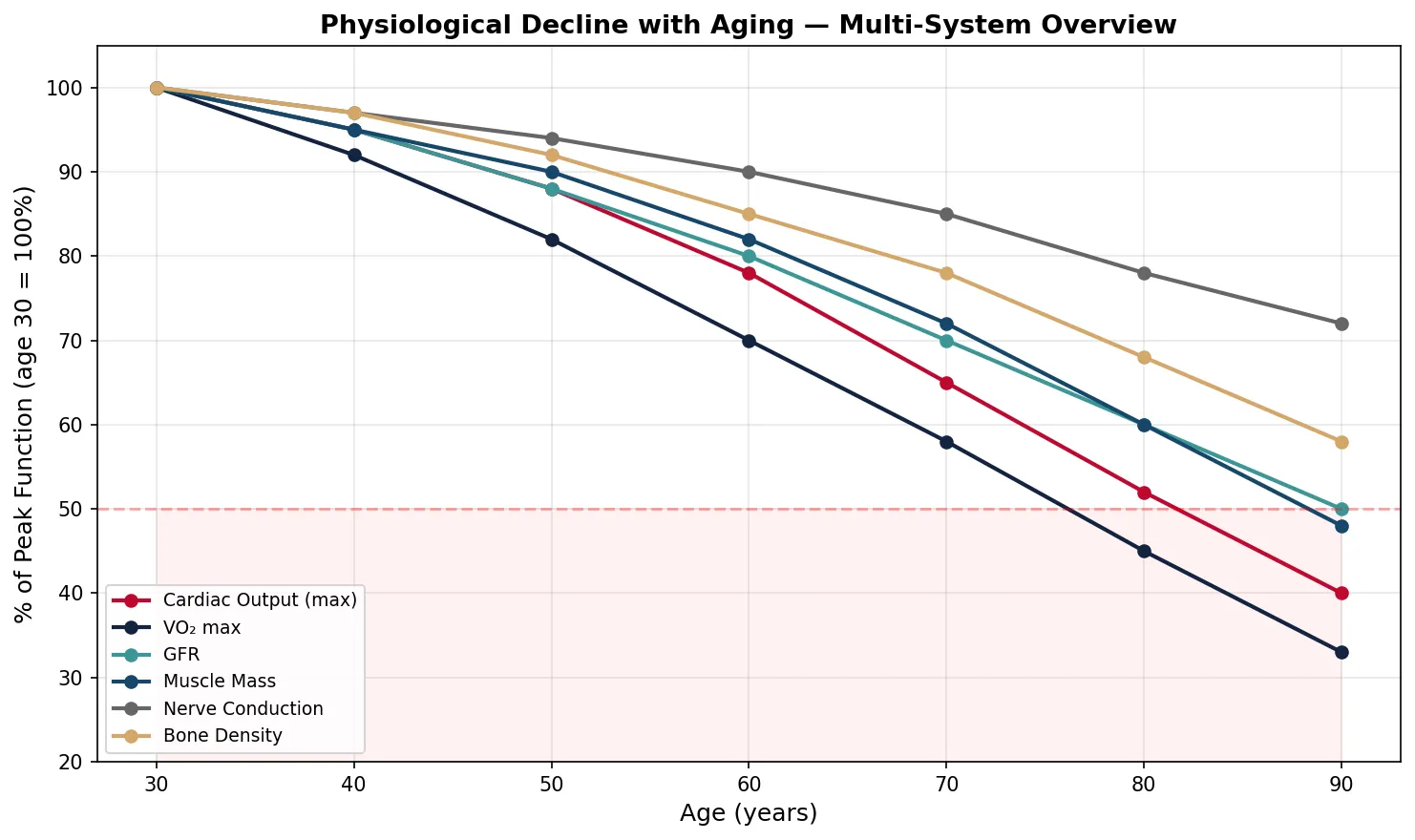
import numpy as np
import matplotlib.pyplot as plt
# Physiological decline with aging (approximate % of peak function remaining)
age = np.array([30, 40, 50, 60, 70, 80, 90])
systems = {
'Cardiac Output (max)': [100, 95, 88, 78, 65, 52, 40],
'VO₂ max': [100, 92, 82, 70, 58, 45, 33],
'GFR': [100, 95, 88, 80, 70, 60, 50],
'Muscle Mass': [100, 95, 90, 82, 72, 60, 48],
'Nerve Conduction': [100, 97, 94, 90, 85, 78, 72],
'Bone Density': [100, 97, 92, 85, 78, 68, 58],
}
fig, ax = plt.subplots(figsize=(10, 6))
colors = ['#BF092F', '#132440', '#3B9797', '#16476A', '#666666', '#D4A76A']
for (name, values), color in zip(systems.items(), colors):
ax.plot(age, values, marker='o', linewidth=2, label=name, color=color)
ax.set_xlabel('Age (years)', fontsize=12)
ax.set_ylabel('% of Peak Function (age 30 = 100%)', fontsize=12)
ax.set_title('Physiological Decline with Aging — Multi-System Overview', fontsize=13, fontweight='bold')
ax.legend(loc='lower left', fontsize=9)
ax.set_ylim(20, 105)
ax.grid(True, alpha=0.3)
ax.axhline(y=50, color='red', linestyle='--', alpha=0.3, label='Critical threshold')
ax.fill_between(age, 0, 50, alpha=0.05, color='red')
plt.tight_layout()
plt.show()
print("Key insight: VO2max and max cardiac output decline fastest; nerve conduction slowest.")
print("Below 50% functional reserve → vulnerability to decompensation with physiological stress.")
Clinical Integration
Clinical physiology brings together every concept from this 12-part series into the practical reality of patient care. Understanding physiology transforms clinicians from protocol-followers into problem-solvers — able to reason from first principles when textbook scenarios don't apply.
Physiological Monitoring
Modern critical care rests on the continuous measurement of physiological variables — each one representing a concept we've studied:
| Monitor | Physiology Measured | Part Reference | Clinical Application |
|---|---|---|---|
| ECG | Cardiac electrophysiology | Part 3 | Arrhythmia detection, ischaemia monitoring, electrolyte effects (K⁺, Ca²⁺) |
| Pulse Oximetry | O₂-Hb dissociation curve | Parts 4, 10 | SpO₂; limitations with CO poisoning, methaemoglobinaemia, poor perfusion |
| Arterial Line | Haemodynamics (SV, CO via pulse contour) | Part 3 | Beat-to-beat BP; arterial waveform analysis; SVV for fluid responsiveness |
| Capnography (ETCO₂) | Ventilation-perfusion, metabolic CO₂ production | Part 4 | Ventilation adequacy; ↓ ETCO₂ = ↓ CO (cardiac arrest); ↑ ETCO₂ = MH |
| CVP / PA catheter | Preload, cardiac function, PCWP | Parts 3, 5 | Distinguish shock types; guide fluid vs vasopressor therapy |
| EEG | Cortical electrical activity | Part 2 | Seizure detection, depth of anaesthesia (BIS), brain death confirmation |
| Urine Output | Renal perfusion, GFR, tubular function | Part 5 | >0.5 mL/kg/h = adequate renal perfusion; oliguria = shock indicator |
Organ System Interactions in Disease
Real clinical scenarios involve cascading failures across systems — demonstrating the interconnectedness of physiology:
The Cardiorenal Syndrome — A Cascade of Organ Failure
A 72-year-old man with chronic heart failure (EF 25%) presents with worsening dyspnoea, bilateral basal crackles, elevated JVP, peripheral oedema, and Cr rising from 1.2 → 2.5 mg/dL over 3 days.
Cascade analysis:
- Heart (Part 3): ↓ CO → ↓ renal perfusion → activates RAAS → Na⁺/H₂O retention → ↑ preload → ↑ venous congestion (elevated CVP transmitted to renal veins → ↓ GFR independent of forward flow)
- Kidneys (Part 5): ↓ GFR → fluid overload → ↑ preload → further cardiac distension (Frank-Starling on descending limb) → ↓ CO → worsening renal function (vicious cycle)
- Lungs (Part 4): ↑ LV filling pressure → ↑ pulmonary capillary hydrostatic pressure → transudative pulmonary oedema → ↓ gas exchange → hypoxia → further myocardial depression
- Acid-base (Part 5): Renal failure → metabolic acidosis → ↓ myocardial contractility → compensatory hyperventilation (Kussmaul breathing)
- Electrolytes: ↓ GFR → ↑ K⁺ → risk of fatal arrhythmia (Part 3 — cardiac action potential dependency on K⁺)
This single case integrates cardiac physiology, renal physiology, respiratory physiology, acid-base balance, and electrolyte homeostasis — demonstrating why physiology must be understood as a system of systems.
Pharmacophysiology Principles
Every drug acts by modifying a physiological process. Understanding the physiology unlocks the pharmacology:
- β-blockers (Part 3): Block β1 receptors → ↓ HR, ↓ contractility → ↓ myocardial O₂ demand. Understanding this explains contraindications: decompensated HF (↓ CO), severe asthma (β2 blockade → bronchoconstriction), bradycardia, heart block.
- ACE inhibitors (Part 5): Block angiotensin I → II conversion → ↓ aldosterone, ↓ vasoconstriction, ↓ cardiac remodelling. Side effects predicted by physiology: hyperkalaemia (↓ aldosterone → ↓ K⁺ excretion), ↓ GFR in renal artery stenosis (AT-II maintains GFR by efferent constriction), cough (↑ bradykinin — ACE also degrades bradykinin).
- Loop diuretics (Part 5): Block Na⁺/K⁺/2Cl⁻ co-transporter in thick ascending limb → massive natriuresis. Understanding tubular physiology predicts: ↓ K⁺ (↑ Na⁺ delivery to collecting duct → ↑ K⁺ secretion), metabolic alkalosis (H⁺/K⁺ exchange with ↓ K⁺), ↓ Ca²⁺ (unlike thiazides, which ↑ Ca²⁺ reabsorption in DCT).
- Insulin (Part 7): Replaces the action of endogenous insulin → ↓ hepatic glucose output, ↑ peripheral glucose uptake, ↓ lipolysis. Understanding insulin physiology predicts: hypoglycaemia (excessive dose or missed meal), hypokalaemia (insulin drives K⁺ into cells — exploited therapeutically in acute hyperkalaemia), weight gain (anabolic hormone).
- Neuromuscular blockers (Part 2): Depolarising (succinylcholine — mimics ACh → sustained depolarisation → block) vs non-depolarising (rocuronium — competitive antagonist at nAChR). Understanding the neuromuscular junction physiology explains: succinylcholine's fasciculations, hyperkalaemia risk in burns/denervation, and why neostigmine reverses non-depolarising agents (↑ ACh at NMJ by inhibiting AChE).
Future Directions in Physiology
- Organ-on-a-chip: Microfluidic devices that recapitulate human organ physiology on a miniature scale — lung-on-chip, heart-on-chip, gut-on-chip — enabling drug testing and disease modelling without animal models
- Digital twins: Computational models of individual patients' physiology, continuously updated with real-time monitoring data, enabling predictive personalised medicine — "what if we give this drug?" simulated before actual administration
- Optogenetics: Light-sensitive ion channels (channelrhodopsins) genetically inserted into specific neurons → precise control of neural circuits with millisecond resolution. Transforming our understanding of brain physiology and offering potential therapies for epilepsy, Parkinson's, and depression.
- Senolytics: Drugs that selectively clear senescent cells (cells that have stopped dividing but remain metabolically active, secreting inflammatory mediators — the "senescence-associated secretory phenotype" or SASP). Early trials suggest potential for reversing age-related decline in multiple organ systems.
- Closed-loop physiological systems: Artificial pancreas (continuous glucose monitor + insulin pump + algorithm) — already FDA-approved; future applications: closed-loop ventilation, closed-loop vasopressor titration in ICU, closed-loop anaesthesia.
Interactive Tool
Use this Clinical Case Integration Report generator to document multi-system clinical cases, connecting pathophysiology across organ systems. Export as Word, Excel, or PDF.
Clinical Case Integration Report
Analyse a clinical case through the lens of integrative physiology. Document the presenting complaint, systems affected, pathophysiology, vitals, and learning points. Download as Word, Excel, or PDF.
Practice Exercises
Conclusion & Series Summary
This final part has demonstrated that clinical medicine is, at its core, applied physiology. The stress response integrates the sympathoadrenal system (Part 2) with the HPA axis (Part 7) and immune modulation (Part 10). Shock states test the limits of cardiovascular compensation (Part 3), renal autoregulation (Part 5), and metabolic adaptation (Part 6). Sepsis reveals how immune defence mechanisms (Part 10) can become self-destructive, damaging lungs (Part 4), kidneys (Part 5), liver (Part 6), brain (Part 2), and the coagulation system (Part 10) simultaneously. Thermoregulation extremes illustrate hypothalamic control (Part 7) and cellular vulnerability. Ageing demonstrates the progressive erosion of every system's reserve capacity.
Throughout this 12-part Physiology Series, we have built understanding from the cellular level (membrane physiology, action potentials) through organ-system physiology (cardiac, respiratory, renal, GI, endocrine, immune, reproductive) to the integrative and clinical level. The unifying theme has been homeostasis — the body's relentless drive to maintain internal stability in a constantly changing environment. When homeostatic mechanisms succeed, we call it health. When they fail, we call it disease. The clinician's role is to understand these mechanisms deeply enough to support them when they falter and to correct them when they fail.